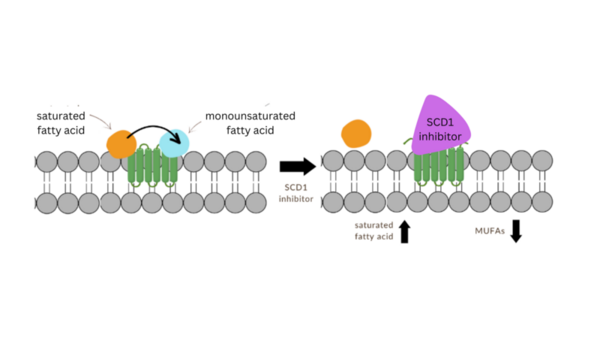
The authors use molecular modeling to test analogs of the stearoyl-coenzyme A desaturase 1 (SCD1) inhibitor MF-438 with implications for future development of Parkinson's disease therapeutics.
Read More...Modeling stearoyl-coenzyme A desaturase 1 inhibitors to ameliorate α-Syn cytotoxicity in Parkinson's disease
The authors use molecular modeling to test analogs of the stearoyl-coenzyme A desaturase 1 (SCD1) inhibitor MF-438 with implications for future development of Parkinson's disease therapeutics.
Read More...Exploring Interactions between PFAS (Per- and Polyfluoroalkyl Substances) and proteins
Here the authors investigated how the "forever chemical" perfluorooctanoic acid binds to bovine serum albumin (BSA) using computational software to simulate its potential impact on essential human plasma proteins. They identify a possible, high-energy binding configuration that could persistently impair protein functions, underscoring the critical need for further research into the long-term health risks of per- and poly-fluoroalkyl substances exposure.
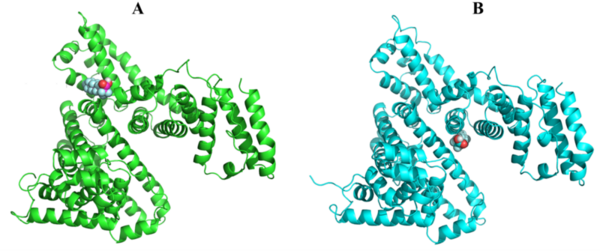
Read More...Homology modeling of clinically-relevant rilpivirine-resistant HIV-RT variants identifies novel rilpivirine analogs with retained binding affinity against NNRTI-resistant HIV mutations
.jpg)
Human immunodeficiency virus (HIV), which affects tens of millions of individuals worldwide, can lead to acquired immunodeficiency syndrome (AIDS). While there is currently no cure for HIV, the development of small molecule antiretroviral agents has greatly improved the prognosis of infected individuals, especially in developed countries. Here, the authors employ homology modeling and molecular docking towards the identification of novel rilpivirine analogs that retain high binding affinity to clinically relevant rilpivirine-resistant mutations of the HIV reverse transcriptase enzyme.
Read More...In silico modeling of emodin’s interactions with serine/threonine kinases and chitosan derivatives

Here, through protein-ligand docking, the authors investigated the effect of the interaction of emodin with serine/threonine kinases, a subclass of kinases that is overexpressed in many cancers, which is implicated in phosphorylation cascades. Through molecular dynamics theyfound that emodin forms favorable interactions with chitosan and chitosan PEG (polyethylene glycol) copolymers, which could aid in loading drugs into nanoparticles (NPs) for targeted delivery to cancerous tissue. Both polymers demonstrated reasonable entrapment efficiencies, which encourages experimental exploration of emodin through targeted drug delivery vehicles and their anticancer activity.
Read More...The Dependence of CO2 Removal Efficiency on its Injection Speed into Water
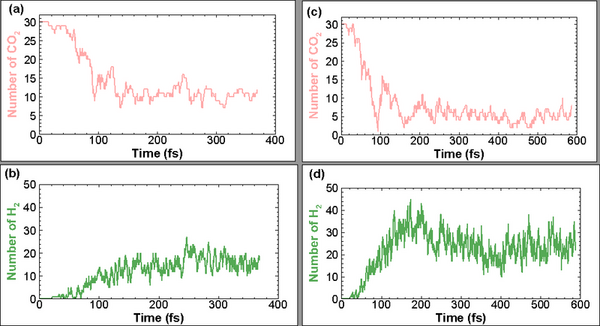
Recent research confirms that climate change, driven by CO2 emissions from burning fossil fuels, poses a significant threat to humanity. In response, authors explore methods to remove CO2 from the atmosphere, including breaking its molecular bonds through high-speed collisions.
Read More...The characterization of quorum sensing trajectories of Vibrio fischeri using longitudinal data analytics
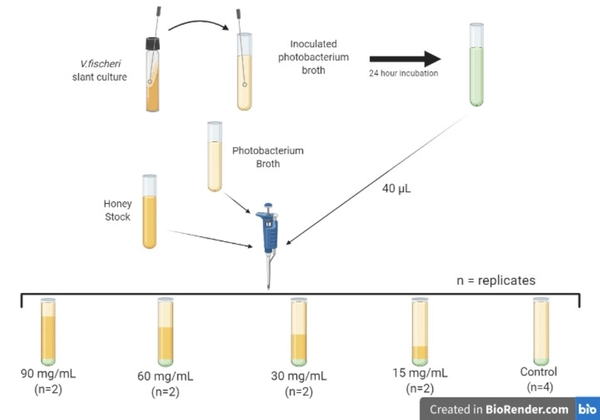
Quorum sensing (QS) is the process in which bacteria recognize and respond to the surrounding cell density, and it can be inhibited by certain antimicrobial substances. This study showed that illumination intensity data is insufficient for evaluating QS activity without proper statistical modeling. It concluded that modeling illumination intensity through time provides a more accurate evaluation of QS activity than conventional cross-sectional analysis.
Read More...Strain-selective in vitro and in silico structure activity relationship (SAR) of N-acyl β-lactam broad spectrum antibiotics
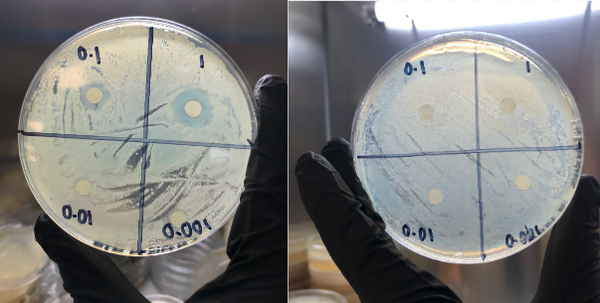
In this study, the authors investigate the antibacterial efficacy of penicillin G and its analogs amoxicillin, carbenicillin, piperacillin, cloxacillin, and ampicillin, against four species of bacteria. Results showed that all six penicillin-type antibiotics inhibit Staphylococcus epidermidis, Escherichia coli, and Neisseria sicca with varying degrees of efficacy but exhibited no inhibition against Bacillus cereus. Penicillin G had the greatest broad-spectrum antibacterial activity with a high radius of inhibition against S. epidermidis, E. coli, and N. sicca.
Read More...Hybrid Quantum-Classical Generative Adversarial Network for synthesizing chemically feasible molecules
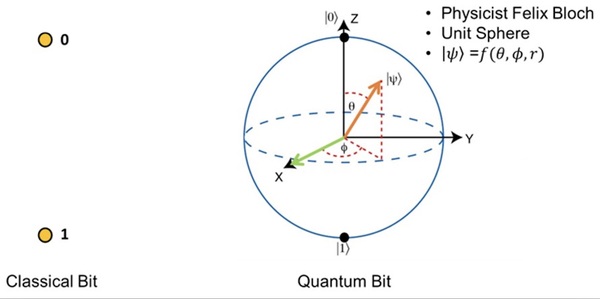
Current drug discovery processes can cost billions of dollars and usually take five to ten years. People have been researching and implementing various computational approaches to search for molecules and compounds from the chemical space, which can be on the order of 1060 molecules. One solution involves deep generative models, which are artificial intelligence models that learn from nonlinear data by modeling the probability distribution of chemical structures and creating similar data points from the trends it identifies. Aiming for faster runtime and greater robustness when analyzing high-dimensional data, we designed and implemented a Hybrid Quantum-Classical Generative Adversarial Network (QGAN) to synthesize molecules.
Read More...Modular mimics of neuroactive alkaloids - design, synthesis, and cholinesterase inhibitory activity of rivastigmine analogs
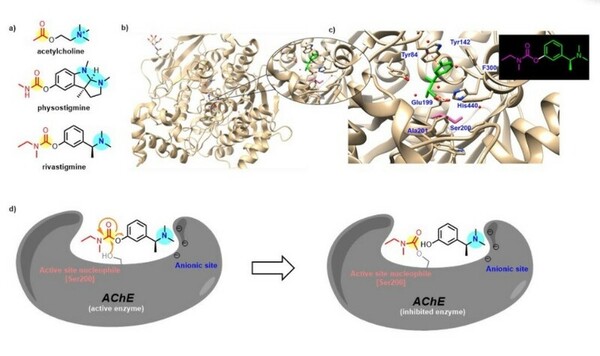
Naturally occurring neuroactive alkaloids are often studied for their potential to treat Neurological diseases. This team of students study Rivastigmine, a potent cholinesterase inhibitor that is a synthetic analog of physostigmine, which comes from the Calabar bean plant Physostigma venenosum. By comparing the effects of optimized synthetic analogs to the naturally occurring alkaloid, they determine the most favorable analog for inhibition of acetylcholinesterase (AChE), the enzyme that breaks down the neurotransmitter acetylcholine (ACh) to terminate neuronal transmission and signaling between synapses.
Read More...Prediction of molecular energy using Coulomb matrix and Graph Neural Network
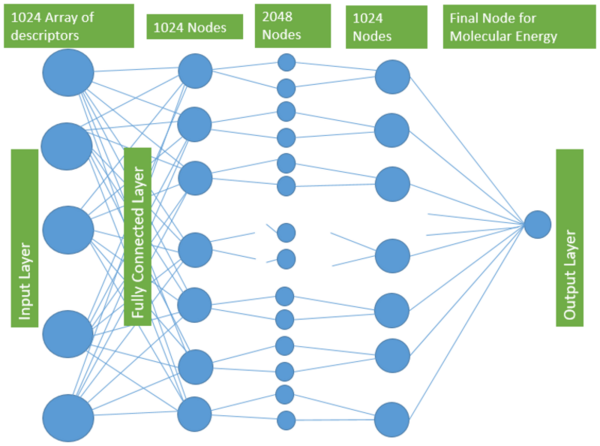
With molecular energy being an integral element to the study of molecules and molecular interactions, computational methods to determine molecular energy are used for the preservation of time and resources. However, these computational methods have high demand for computer resources, limiting their widespread feasibility. The authors of this study employed machine learning to address this disadvantage, utilizing neural networks trained on different representations of molecules to predict molecular properties without the requirement of computationally-intensive processing. In their findings, the authors determined the Feedforward Neural Network, trained by two separate models, as capable of predicting molecular energy with limited prediction error.
Read More...