
In this study, the authors address the concerns of heavy metal contamination in industrial and feedlot water waste. They test whether added probiotics are capable of taking up heavy metals in water to attenuate pollution.
Read More...Probiotic biosorption as a way to remove heavy metal in seawater
In this study, the authors address the concerns of heavy metal contamination in industrial and feedlot water waste. They test whether added probiotics are capable of taking up heavy metals in water to attenuate pollution.
Read More...Transcriptional Regulators are Upregulated in the Substantia Nigra of Parkinson’s Disease Patients
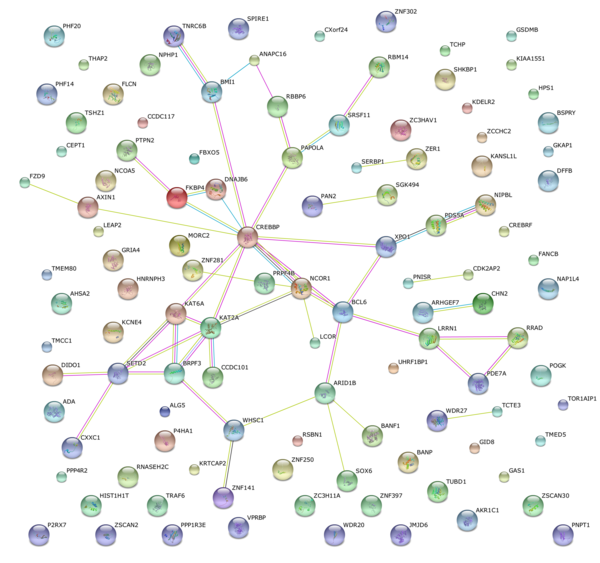
This article investigates differences in gene expression in the brains of patients with and without Parkinson's disease. The authors identify a crucial transcriptional regulator may be a relevant target for future therapeutic treatment for Parkinson's disease.
Read More...Extending Einstein’s elevator thought experiment to multiple spatial dimensions at the Luxor Hotel & Casino
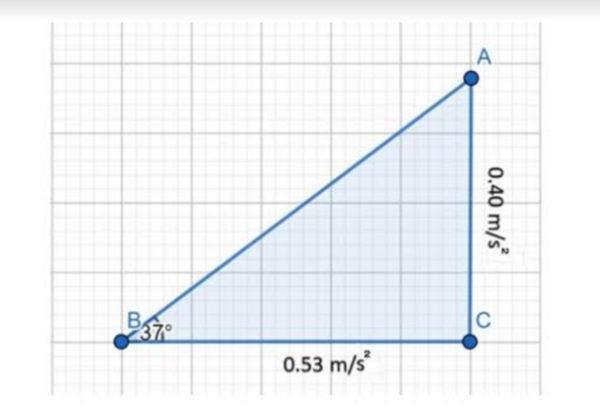
In this study, the authors conduct a series of experiments within an elevator traveling on an angle to determine if Einstein's Equivalency Principle and motion vector decomposition can be used to calculate the angle of inclination.
Read More...Investigating ecosystem resiliency in different flood zones of south Brooklyn, New York
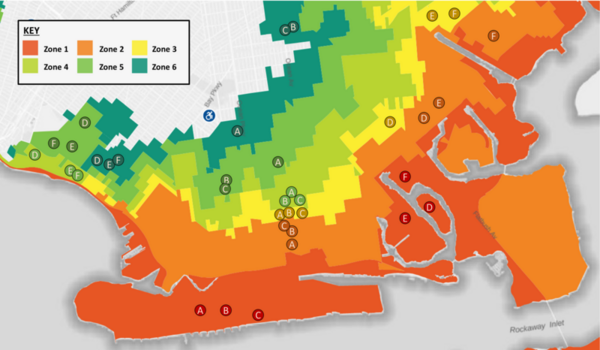
With climate change and rising sea levels, south Brooklyn is exposed to massive flooding and intense precipitation. Previous research discovered that flooding shifts plant species distribution, decreases soil pH, and increases salt concentration, nitrogen, phosphorus, and potassium levels. The authors predicted a decreasing trend from Zone 1 to 6: high-pH, high-salt, and high-nutrients in more flood-prone areas to low-pH, low-salt, and low-nutrient in less flood-prone regions. They performed DNA barcoding to identify plant species inhabiting flood zones with expectations of decreasing salt tolerance and moisture uptake by plants' soil from Zones 1-6. Furthermore, they predicted an increase in invasive species, ultimately resulting in a decrease in biodiversity. After barcoding, they researched existing information regarding invasiveness, ideal soil, pH tolerance, and salt tolerance. They performed soil analyses to identify pH, nitrogen (N), phosphorus (P), and potassium (K) levels. For N and P levels, we discovered a general decreasing trend from Zone 1 to 6 with low and moderate statistical significance respectively. Previous studies found that soil moisture can increase N and P uptake, helping plants adopt efficient resource-use strategies and reduce water stress from flooding. Although characteristics of plants were distributed throughout all zones, demonstrating overall diversity, the soil analyses hinted at the possibility of a rising trend of plants adapting to the increase in flooding. Future expansive research is needed to comprehensively map these trends. Ultimately, investigating trends between flood zones and the prevalence of different species will assist in guiding solutions to weathering climate change and protecting biodiversity in Brooklyn.
Read More...Recognition of animal body parts via supervised learning
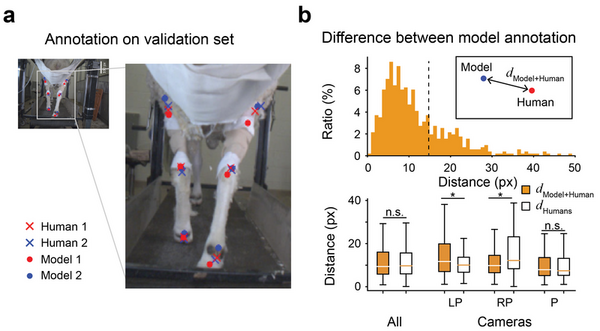
The application of machine learning techniques has facilitated the automatic annotation of behavior in video sequences, offering a promising approach for ethological studies by reducing the manual effort required for annotating each video frame. Nevertheless, before solely relying on machine-generated annotations, it is essential to evaluate the accuracy of these annotations to ensure their reliability and applicability. While it is conventionally accepted that there cannot be a perfect annotation, the degree of error associated with machine-generated annotations should be commensurate with the error between different human annotators. We hypothesized that machine learning supervised with adequate human annotations would be able to accurately predict body parts from video sequences. Here, we conducted a comparative analysis of the quality of annotations generated by humans and machines for the body parts of sheep during treadmill walking. For human annotation, two annotators manually labeled six body parts of sheep in 300 frames. To generate machine annotations, we employed the state-of-the-art pose-estimating library, DeepLabCut, which was trained using the frames annotated by human annotators. As expected, the human annotations demonstrated high consistency between annotators. Notably, the machine learning algorithm also generated accurate predictions, with errors comparable to those between humans. We also observed that abnormal annotations with a high error could be revised by introducing Kalman Filtering, which interpolates the trajectory of body parts over the time series, enhancing robustness. Our results suggest that conventional transfer learning methods can generate behavior annotations as accurate as those made by humans, presenting great potential for further research.
Read More...Refinement of Single Nucleotide Polymorphisms of Atopic Dermatitis related Filaggrin through R packages

In the United States, there are currently 17.8 million affected by atopic dermatitis (AD), commonly known as eczema. It is characterized by itching and skin inflammation. AD patients are at higher risk for infections, depression, cancer, and suicide. Genetics, environment, and stress are some of the causes of the disease. With the rise of personalized medicine and the acceptance of gene-editing technologies, AD-related variations need to be identified for treatment. Genome-wide association studies (GWAS) have associated the Filaggrin (FLG) gene with AD but have not identified specific problematic single nucleotide polymorphisms (SNPs). This research aimed to refine known SNPs of FLG for gene editing technologies to establish a causal link between specific SNPs and the diseases and to target the polymorphisms. The research utilized R and its Bioconductor packages to refine data from the National Center for Biotechnology Information's (NCBI's) Variation Viewer. The algorithm filtered the dataset by coding regions and conserved domains. The algorithm also removed synonymous variations and treated non-synonymous, frameshift, and nonsense separately. The non-synonymous variations were refined and ordered by the BLOSUM62 substitution matrix. Overall, the analysis removed 96.65% of data, which was redundant or not the focus of the research and ordered the remaining relevant data by impact. The code for the project can also be repurposed as a tool for other diseases. The research can help solve GWAS's imprecise identification challenge. This research is the first step in providing the refined databases required for gene-editing treatment.
Read More...The Development of a Superhydrophobic Surface Using Electrolytic Deposition & Polymer Chains Precipitation
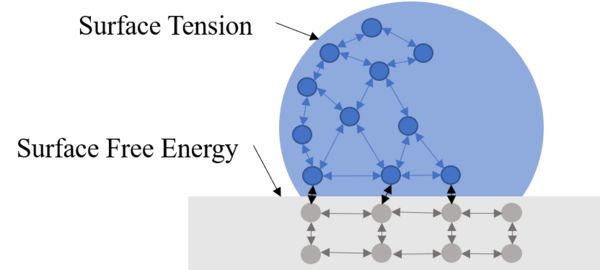
In this study, the authors were interested in developing a hydrophobic surface that will extend the lifespan of metals by reducing water exposure and other damage. The used a zinc coating on steel to pursue this effort.
Read More...Predicting the Instance of Breast Cancer within Patients using a Convolutional Neural Network
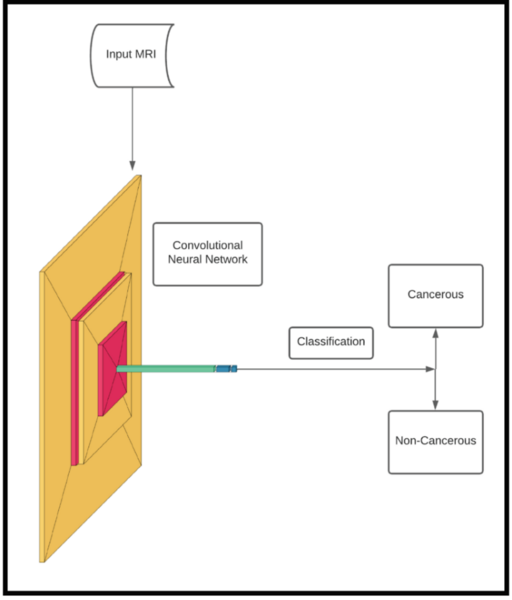
Using a convolution neural network, these authors show machine learning can clinically diagnose breast cancer with high accuracy.
Read More...Uncovering mirror neurons’ molecular identity by single cell transcriptomics and microarray analysis
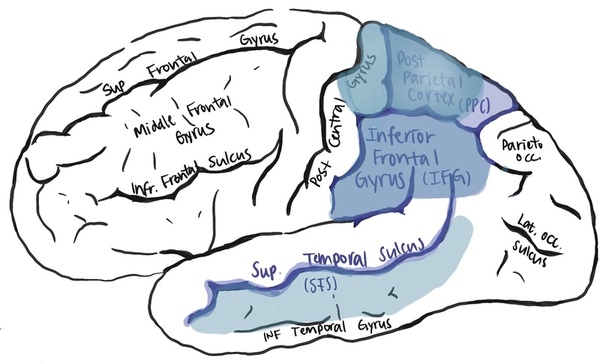
In this study, the authors use bioinformatic approaches to characterize the mirror neurons, which are active when performing and seeing certain actions. They also investigated whether mirror neuron impairment was connected to neural degenerative diseases and psychiatric disorders.
Read More...Using explainable artificial intelligence to identify patient-specific breast cancer subtypes
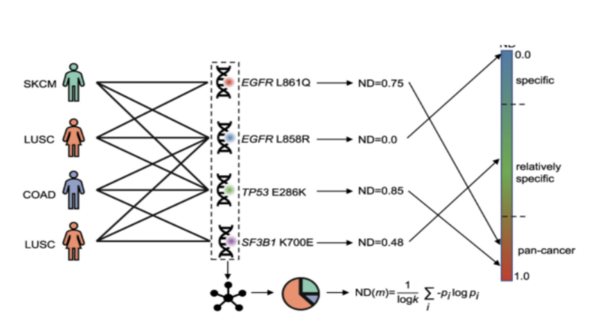
Breast cancer is the most common cancer in women, with approximately 300,000 diagnosed with breast cancer in 2023. It ranks second in cancer-related deaths for women, after lung cancer with nearly 50,000 deaths. Scientists have identified important genetic mutations in genes like BRCA1 and BRCA2 that lead to the development of breast cancer, but previous studies were limited as they focused on specific populations. To overcome limitations, diverse populations and powerful statistical methods like genome-wide association studies and whole-genome sequencing are needed. Explainable artificial intelligence (XAI) can be used in oncology and breast cancer research to overcome these limitations of specificity as it can analyze datasets of diagnosed patients by providing interpretable explanations for identified patterns and predictions. This project aims to achieve technological and medicinal goals by using advanced algorithms to identify breast cancer subtypes for faster diagnoses. Multiple methods were utilized to develop an efficient algorithm. We hypothesized that an XAI approach would be best as it can assign scores to genes, specifically with a 90% success rate. To test that, we ran multiple trials utilizing XAI methods through the identification of class-specific and patient-specific key genes. We found that the study demonstrated a pipeline that combines multiple XAI techniques to identify potential biomarker genes for breast cancer with a 95% success rate.
Read More...Search articles by title, author name, or tags