The authors use computational methods to compare tau acetylation to the better studied tau phosphorylation in Alzheimer's disease and then design and computationally test a new drug to prevent abnormal post-translational modifications of tau.
Berberine is a natural quaternary alkaloid that has anti-microbial and anti-cancer effects. This compound can bind to Guanine Quadruplex (G4) DNA secondary complexes to help inhibit cancer cell proliferation. In this study, the authors investigate whether incorporating large aromatic rings helps to stabilize berberine-G4 interactions.
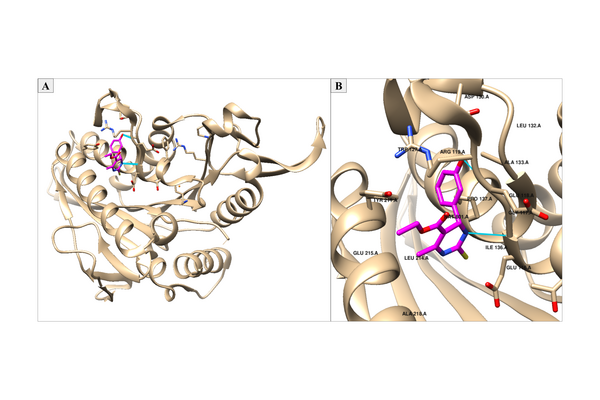
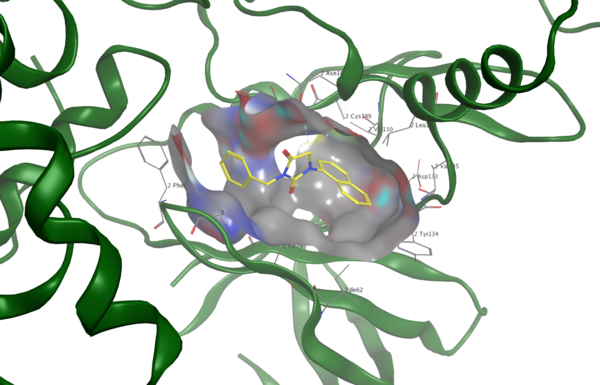
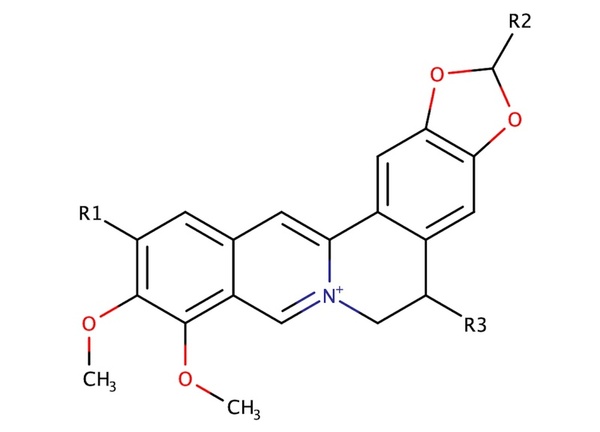
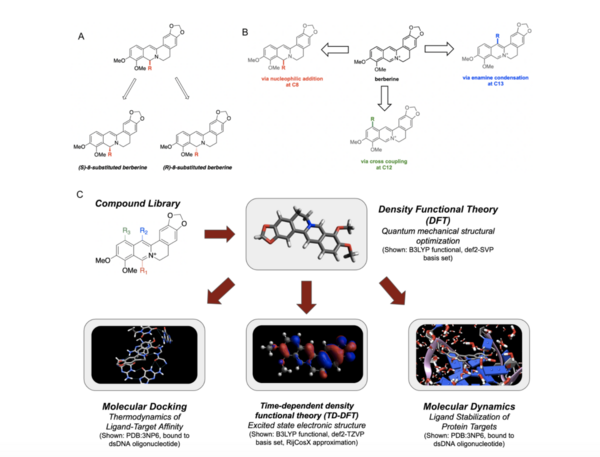
Berberine, a natural product alkaloid, and its analogs have a wide range of medicinal properties, including antibacterial and anticancer effects. Here, the authors explored a library of alkyl or aryl berberine analogs to probe binding to double-stranded and G-quadruplex DNA. They determined that the nature of the substituent, the position of the substituent, and the nucleic acid target affect the free energy of binding of berberine analogs to DNA and G-quadruplex DNA, however berberine analogs did not result in net stabilization of G-quadruplex DNA.
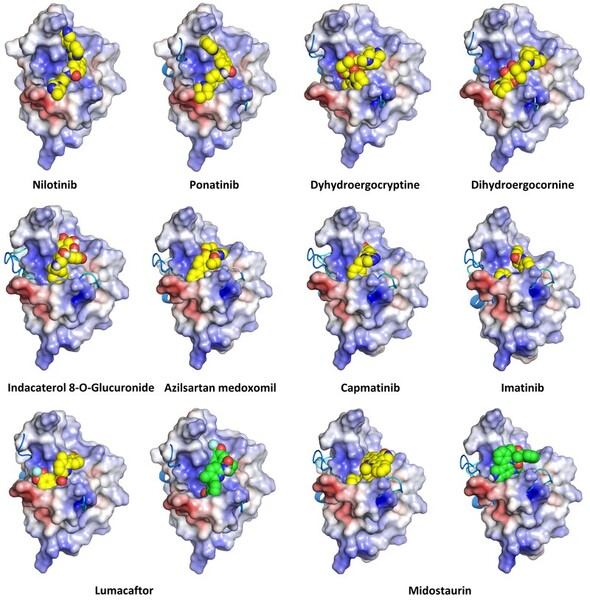
Here, through protein-ligand docking, the authors investigated the effect of the interaction of emodin with serine/threonine kinases, a subclass of kinases that is overexpressed in many cancers, which is implicated in phosphorylation cascades. Through molecular dynamics theyfound that emodin forms favorable interactions with chitosan and chitosan PEG (polyethylene glycol) copolymers, which could aid in loading drugs into nanoparticles (NPs) for targeted delivery to cancerous tissue. Both polymers demonstrated reasonable entrapment efficiencies, which encourages experimental exploration of emodin through targeted drug delivery vehicles and their anticancer activity.
Molecules which bind to proteins that aggregate abnormally in neurodegenerative diseases could be promising drugs for these diseases. In this study, Zhang, Wu, Zhang, and Dang simulate the binding behavior of various molecules to screen for candidates which could be promising candidates for drug development.
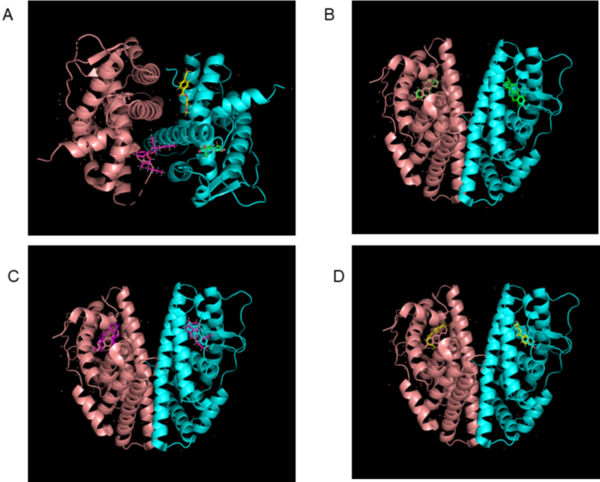
Anticholinergics are used in treating asthma, a chronic inflammation of the airways. These drugs block human M1 and M2 muscarinic acetylcholine receptors, inhibiting bronchoconstriction. However, studies have reported complications of anticholinergic usage, such as exacerbated eosinophil production and worsened urinary retention. Modification of known anticholinergics using bioisosteric replacements to increase efficacy could potentially minimize these complications. The present study focuses on identifying viable analogs of anticholinergics to improve binding energy to the receptors compared to current treatment options. Glycopyrrolate (G), ipratropium (IB), and tiotropium bromide (TB) were chosen as parent drugs of interest, due to the presence of common functional groups within the molecules, specifically esters and alcohols. Docking score analysis via AutoDock Vina was used to evaluate the binding energy between drug analogs and the muscarinic acetylcholine receptors. The final results suggest that G-A3, IB-A3, and TB-A1 are the most viable analogs, as binding energy was improved when compared to the parent drug. G-A4, IB-A4, IB-A5, TB-A3, and TB-A4 are also potential candidates, although there were slight regressions in binding energy to both muscarinic receptors for these analogs. By researching the effects of bioisosteric replacements of current anticholinergics, it is evident that there is a potential to provide asthmatics with more effective treatment options.
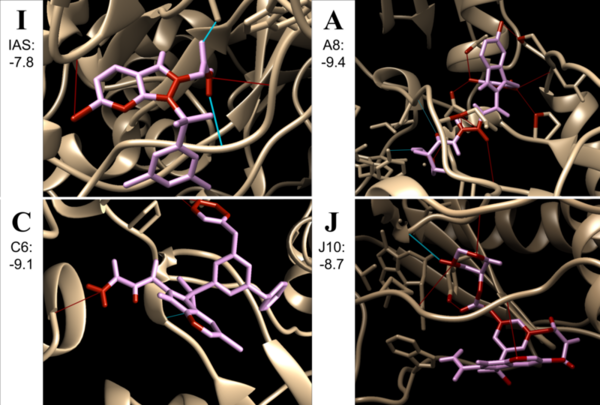
Non-nucleoside reverse transcriptase inhibitors (NNRTIs) are allosteric inhibitors that bind to the HIV reverse transcriptase and prevent replication. Indolyl aryl sulfones (IAS) and IAS derivatives have been found to be highly effective against mutant strains of HIV-1 reverse transcriptase. Here, we analyzed molecules designed using aryl sulfone scaffolds paired to cyclic compounds as potential NNRTIs through the computational design and docking of 100 novel NNRTI candidates. Moreover, we explored the specific combinations of functional groups and aryl sulfones that resulted in the NNRTI candidates with the strongest binding affinity while testing all compounds for carcinogenicity. We hypothesized that the combination of an IAS scaffold and pyrimidine would produce the compounds with the best binding affinity. Our hypothesis was correct as the series of molecules with an IAS scaffold and pyrimidine exhibited the best average binding affinity. Additionally, this study found 32 molecules designed in this procedure with higher or equal binding affinities to the previously successful IAS derivative 5-bromo-3-[(3,5-dimethylphenyl)sulfonyl]indole-2-carboxyamide when docked to HIV-1 reverse transcriptase.
In this study, the authors design a series of new biaryl small molecules to target and block the binding pocket of the enzyme dihydropteroate synthase, which is important for prokaryotic biosynthesis of folic acid and could serve as better antimicrobial compounds.
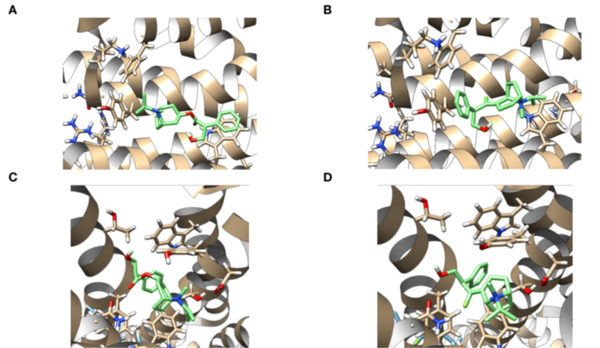
As cancer continues to take millions of lives worldwide, the need to create effective therapeutics for the disease persists. The kinesin Eg5 assembly motor protein is a promising target for cancer therapeutics as inhibition of this protein leads to cell cycle arrest. Monastrol, a small dihydropyrimidine-based molecule capable of inhibiting the kinesin Eg5 function, has attracted the attention of medicinal chemists with its potency, affinity, and specificity to the highly targeted loop5/α2/α3 allosteric binding pocket. In this work, we employed high-throughput virtual screening (HTVS) to identify potential small molecule Eg5 inhibitors from a designed set of novel dihydropyrimidine analogs structurally similar to monastrol.

.jpeg)