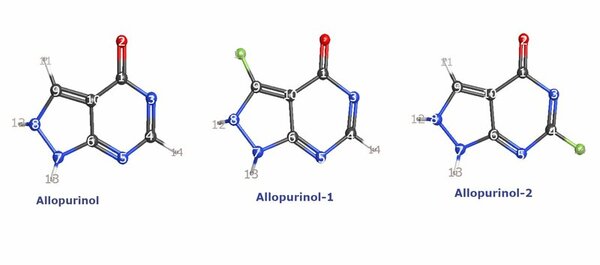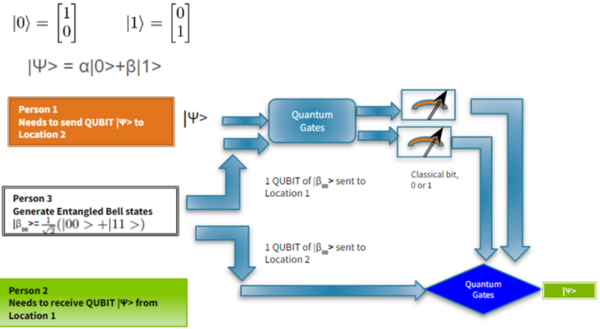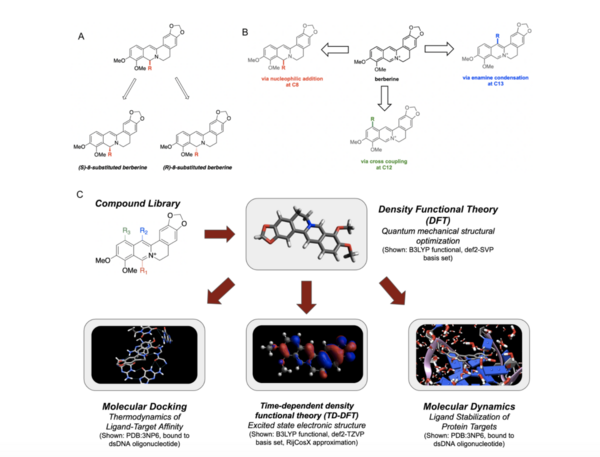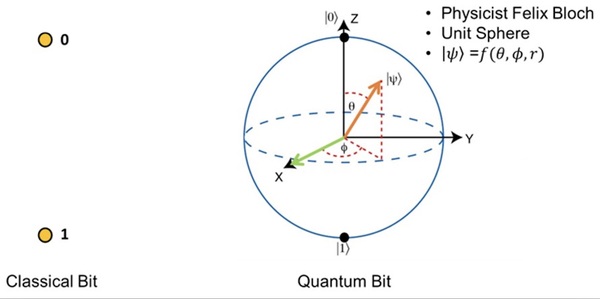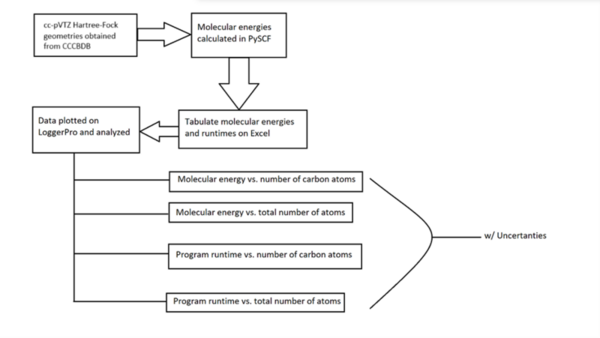
It's time-consuming to complete the calculations that are used to study nuclear reactions and energy. To uncover which computational chemistry tools are useful for this challenge, Pan, Vaiyakarnam, Li, and McMahan investigated whether the Python-based Simulations of Chemistry Framework’s Hartree-Fock (PySCF) method is an efficient and accurate way to assess alkane molecules.
Read More...
.jpg)