Impact of carbon number and atom number on cc-pVTZ Hartree-Fock Energy and program runtime of alkanes
(1) The Quarry Lane School, (2) Monta Vista High School, (3) Aspiring Scholars Directed Research Program
https://doi.org/10.59720/23-130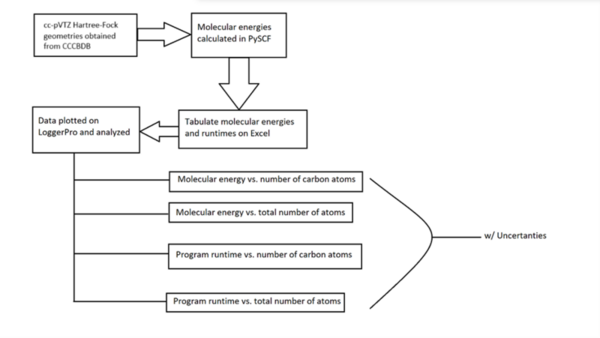
(Pan, Vaiyakarnam, and Li contributed equally to the work) The ground state energy of a molecule can be used in many aspects of chemistry, such as predicting energies produced or absorbed in reactions and determining the stability of the molecules and their analogs. However, different methods to compute these energies have different accuracies and speeds. We aimed to compare one of these methods, the Python-based Simulations of Chemistry Framework’s Hartree-Fock (PySCF) method, with established values provided by the Computational Chemistry Comparison and Benchmark Database (CCCBDB), a reliable peer-reviewed database organized by the United States government. We also sought to assess relationships between the structure of each alkane and the runtime of the PySCF program. Molecular geometries from the CCCBDB were taken for straight- chain alkanes with 1-10 carbon atoms. Using the PySCF Hartree-Fock (HF) method, ground state energies were calculated for these alkanes using triple-zeta (cc-pVTZ) basis sets. These energies were then compared to established HF/cc-pVTZ data energies of the same alkanes in the CCCBDB. We hypothesized that the ground state energy would increase linearly and that the runtime of the program would increase quadratically as both the number of carbon atoms and total atoms increased. The data supported our hypotheses – the ground state energy had a negative linear correlation with the number of carbon atoms and the total number of atoms (r = -1.000), while the runtime had a quadratic correlation with the number of atoms (R2 = 0.9998). The PySCF data also agreed with the CCCBDB data, indicating that PySCF is both efficient and accurate as a computational chemistry software, and can be tested in future experiments with larger organic molecules such as pharmaceutical candidates.
This article has been tagged with: