Sequence accessibility is an important factor affecting gene expression. Sequence accessibility or openness impacts the likelihood that a gene is transcribed and translated into a protein and performs functions and manifests traits. There are many potential factors that affect the accessibility of a gene. In this study, our hypothesis was that the content of nucleotides in a genetic sequence predicts its accessibility. Using a machine learning linear regression model, we studied the relationship between nucleotide content and accessibility.
In this study, the authors developed a model named DNA Sequence Embedding Network (DNA-SEnet) to classify DNA-asthma associations using their genomic patterns.
In the United States, there are currently 17.8 million affected by atopic dermatitis (AD), commonly known as eczema. It is characterized by itching and skin inflammation. AD patients are at higher risk for infections, depression, cancer, and suicide. Genetics, environment, and stress are some of the causes of the disease. With the rise of personalized medicine and the acceptance of gene-editing technologies, AD-related variations need to be identified for treatment. Genome-wide association studies (GWAS) have associated the Filaggrin (FLG) gene with AD but have not identified specific problematic single nucleotide polymorphisms (SNPs). This research aimed to refine known SNPs of FLG for gene editing technologies to establish a causal link between specific SNPs and the diseases and to target the polymorphisms. The research utilized R and its Bioconductor packages to refine data from the National Center for Biotechnology Information's (NCBI's) Variation Viewer. The algorithm filtered the dataset by coding regions and conserved domains. The algorithm also removed synonymous variations and treated non-synonymous, frameshift, and nonsense separately. The non-synonymous variations were refined and ordered by the BLOSUM62 substitution matrix. Overall, the analysis removed 96.65% of data, which was redundant or not the focus of the research and ordered the remaining relevant data by impact. The code for the project can also be repurposed as a tool for other diseases. The research can help solve GWAS's imprecise identification challenge. This research is the first step in providing the refined databases required for gene-editing treatment.
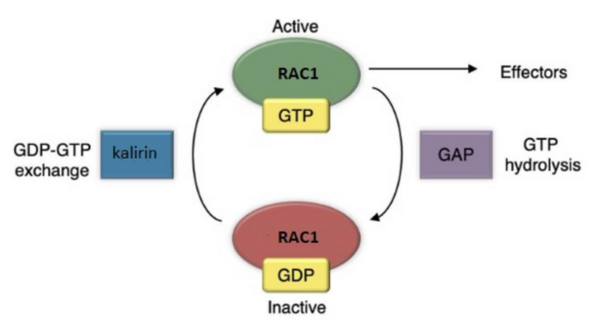
Kalirin is a guanine nucleotide exchange factor (GEF) for the GTPase RAC1, linked to schizophrenia and Alzheimer’s Disease. It plays a crucial role in synaptic plasticity by regulating dendritic spine formation and actin cytoskeleton remodeling, which are essential for creating new synapses. Authors developed two novel compounds targeting kalirin, confirming that predictive modeling can indicate biological activity.
This study sought to determine if there is an association between the single nucleotide polymorphism rs7528684 of the Fc receptor-like-3 (FCRL3) gene and asthma or allergic rhinitis (AR). Based on previous studies in an Asian population, we hypothesized that participants with an AA genotype of FCRL3 would be more likely to have asthma and/or allergic rhinitis. To test the hypothesis, surveys were administered to participants, and genotyping was performed on spit samples via PCR, restriction digest, and gel electrophoresis.
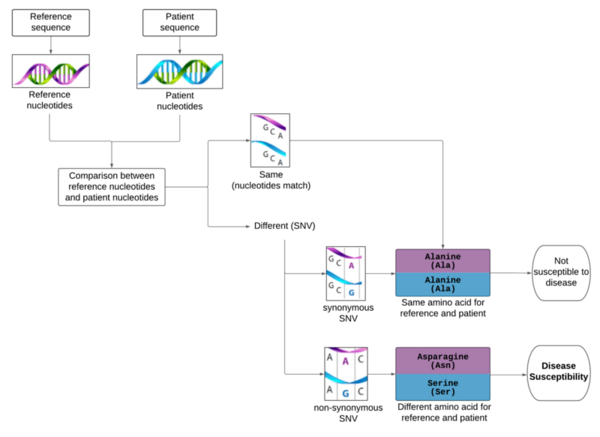
Although the 5-year survival rate for colorectal cancer is below 10%, it increases to greater than 90% if it is diagnosed early. We hypothesized from our research that analyzing non-synonymous single nucleotide variants (SNVs) in a patient's exome sequence would be an indicator for high genetic risk of developing colorectal cancer.
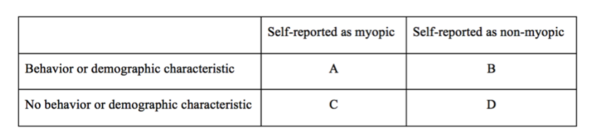
The goal of this project was to assess the relationships among low myopia, behavioral and demographic factors, and a single-nucleotide polymorphism (SNP) in the TGFβ1 gene.
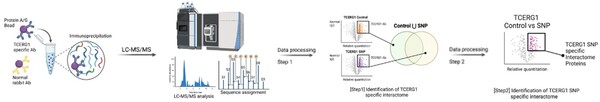
The authors assess a genetic variant within a well-known interaction partner of huntingtin that has been linked to modifying the age of onset of Huntington's disease.
Here, seeking to understand a possible cause of the declining popluations of Brood X cicadas in Ohio and Indiana, the authors investigated the presence of Wolbachia, an inherited bacterial symbiont that lives in the reproductive cells of approximately 60% of insect species in these cicadas. Following their screening of one-hundred 17-year periodical cicadas, they only identified the presence of Wolbachia infection in less than 2%, suggesting that while Wolbachia can infect cicadas it appears uncommon in the Brood X cicadas they surveyed.
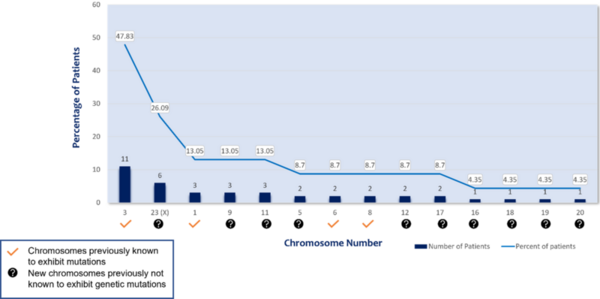
Uveal melanoma (UM) is a rare subtype of melanoma but the most frequent primary cancer of the eye in adults. The goal of this study was to research the genetic causes of UM through a comprehensive frequency analysis of base-pair mismatches in patient genomes. Results showed a total of 130 genetic mutations, including seven recurrent mutations, with most mutations occurring in chromosomes 3 and X. Recurrent mutations varied from 8.7% to 17.39% occurrence in the UM patient sample, with all mutations identified as missense. These findings suggest that UM is a recessive heterogeneous disease with selective homozygous mutations. Notably, this study has potential wider significance because the seven genes targeted by recurrent mutations are also involved in other cancers.