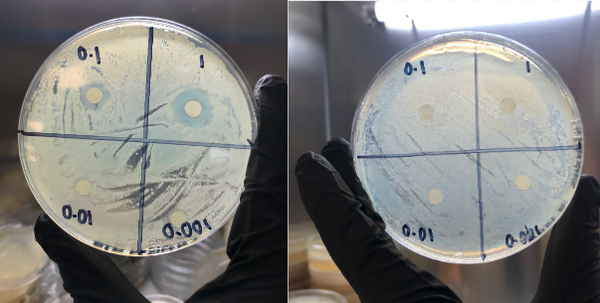
In this study, the authors investigate the antibacterial efficacy of penicillin G and its analogs amoxicillin, carbenicillin, piperacillin, cloxacillin, and ampicillin, against four species of bacteria. Results showed that all six penicillin-type antibiotics inhibit Staphylococcus epidermidis, Escherichia coli, and Neisseria sicca with varying degrees of efficacy but exhibited no inhibition against Bacillus cereus. Penicillin G had the greatest broad-spectrum antibacterial activity with a high radius of inhibition against S. epidermidis, E. coli, and N. sicca.
Read More...