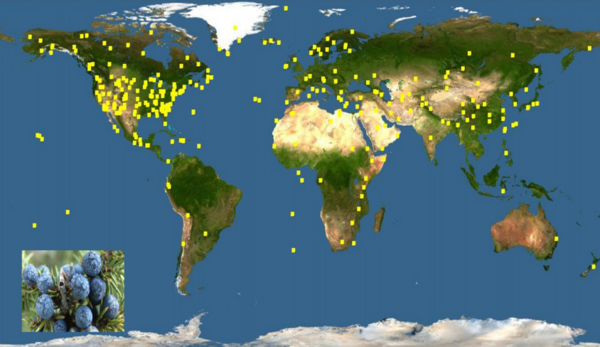
Many species of trees are distributed widely around the world, though not always in a way that makes immediate sense. The authors here use genetic information to help explain the geographic distribution of various conifer species throughout the world.
Although the United States maintains millions of square kilometers of nature reserves to protect the biodiversity of the specimens living there, little is known about how confining these species within designated protected lands influences the genetic variation required for a healthy population. In this study, the authors sequenced genetic barcodes of insects from a recently established nature reserve, the Southwestern Riverside County Multi-Species Reserve (SWRCMSR), and a non-protected area, the Mt. San Jacinto College (MSJC) Menifee campus, to compare the genetic variation between the two populations. Their results demonstrated that the midge fly population from the SWRCMSR had fewer unique DNA barcode sequence changes than the MSJC population, indicating that the comparatively younger nature reserve's population had likely not yet established its own unique genetic drift changes.
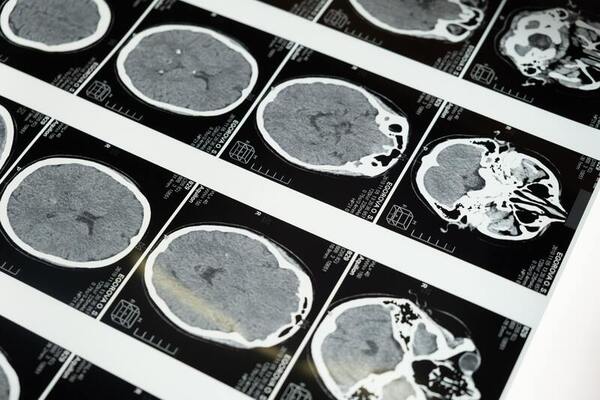
Glioblastoma Multiforme (GBM) is the most malignant brain tumor with the highest fraction of genome alterations (FGA), manifesting poor disease-free status (DFS) and overall survival (OS). We explored The Cancer Genome Atlas (TCGA) and cBioportal public dataset- Firehose legacy GBM to study DNA repair genes Activating Signal Cointegrator 1 Complex Subunit 3 (ASCC3) and Alpha-Ketoglutarate-Dependent Dioxygenase AlkB Homolog 3 (ALKBH3). To test our hypothesis that these genes have correlations with FGA and can better determine prognosis and survival, we sorted the dataset to arrive at 254 patients. Analyzing using RStudio, both ASCC3 and ALKBH3 demonstrated hypomethylation in 82.3% and 61.8% of patients, respectively. Interestingly, low mRNA expression was observed in both these genes. We further conducted correlation tests between both methylation and mRNA expression of these genes with FGA. ASCC3 was found to be negatively correlated, while ALKBH3 was found to be positively correlated, potentially indicating contrasting dysregulation of these two genes. Prognostic analysis showed the following: ASCC3 hypomethylation is significant with DFS and high ASCC3 mRNA expression to be significant with OS, demonstrating ASCC3’s potential as disease prediction marker.
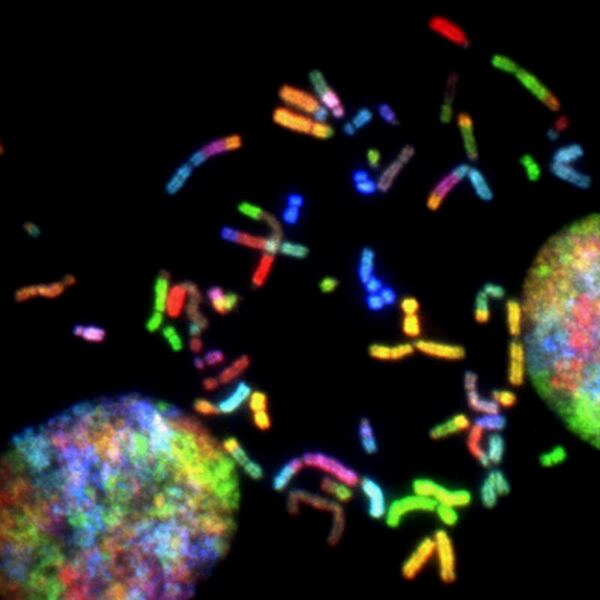
In this study, the authors identify transcripts and gene networks that are changed after infection with the Middle East Respiratory Syndrome-related coronavirus (MERS-CoV).
In the United States, there are currently 17.8 million affected by atopic dermatitis (AD), commonly known as eczema. It is characterized by itching and skin inflammation. AD patients are at higher risk for infections, depression, cancer, and suicide. Genetics, environment, and stress are some of the causes of the disease. With the rise of personalized medicine and the acceptance of gene-editing technologies, AD-related variations need to be identified for treatment. Genome-wide association studies (GWAS) have associated the Filaggrin (FLG) gene with AD but have not identified specific problematic single nucleotide polymorphisms (SNPs). This research aimed to refine known SNPs of FLG for gene editing technologies to establish a causal link between specific SNPs and the diseases and to target the polymorphisms. The research utilized R and its Bioconductor packages to refine data from the National Center for Biotechnology Information's (NCBI's) Variation Viewer. The algorithm filtered the dataset by coding regions and conserved domains. The algorithm also removed synonymous variations and treated non-synonymous, frameshift, and nonsense separately. The non-synonymous variations were refined and ordered by the BLOSUM62 substitution matrix. Overall, the analysis removed 96.65% of data, which was redundant or not the focus of the research and ordered the remaining relevant data by impact. The code for the project can also be repurposed as a tool for other diseases. The research can help solve GWAS's imprecise identification challenge. This research is the first step in providing the refined databases required for gene-editing treatment.
Here the authors sought to better understand glioma, cancer that occurs in the glial cells of the brain with gene expression profile analysis. They considered the expression of complement system genes across the transcriptional and IDH-mutational subtypes of low-grade glioma and glioblastoma. Based on their results of their differential gene expression analysis, they found that outcomes vary across different glioma subtypes, with evidence suggesting that categorization of the transcriptional subtypes could help inform treatment by providing an expectation for treatment responses.
Cancer is often caused by improper function of a few proteins, and sometimes it takes only a few proteins to malfunction to cause drastic changes in cells. Here the authors look at the genes that were mutated in patients with a type of pancreatic cancer to identify proteins that are important in causing cancer. They also determined which proteins currently lack effective treatment, and suggest that certain proteins (named KRAS, CDKN2A, and RBBP8) are the most important candidates for developing drugs to treat pancreatic cancer.
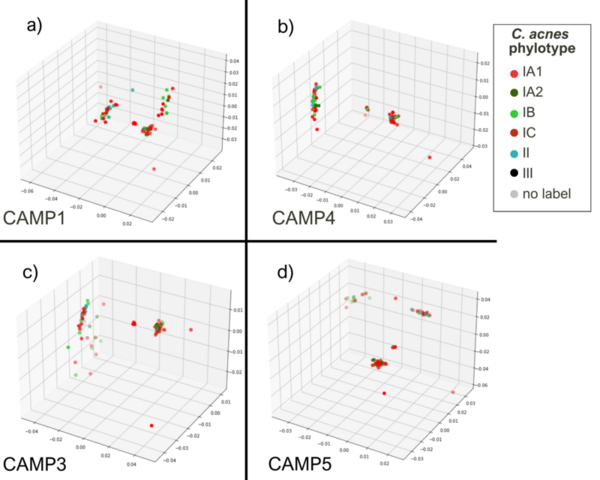
Cutibacterium acnes is a bacterium believed to play an important role in the pathogenesis of common skin diseases such as acne vulgaris. Currently, acne is known to be associated with strains from the type IA1 and IC clades of C. acnes, while those from the type IA2, IB, II, and III phylogroups are associated with skin health. This is the first study to explore the sequence space of individual gene products of different C. acnes phylogroups. Our analysis compared the sequence space topology of virulence factors to proteins with unknown functions and housekeeping proteins. We hypothesized that sequence space features of virulence factors are different from housekeeping protein features, which potentially provides an avenue to deduce unknown proteins’ functions. This proposition should be confirmed based on further experimental outcomes. A notable similarity in the sequence spaces’ topological features of previously known as housekeeping proteins encoded by recA and guaA genes to ‘putative virulence’ genes camp2 and tly was observed. Our research suggests further investigation of recA and guaA’s potential virulence properties to better understand acne pathogenesis and develop more targeted acne treatments.

%20(1).png)