While cephalopods play significant roles in both ecosystems and medical research, there is currently no assembled genome. In an attempt to sequence the Sepia bandensis genome, it was found that there was inhibition from the organism during DNA extraction, resulting in PCR failure. In this study, researchers tested the hypothesis that S. bandensis ink inhibits PCR. They then assessed the impact of ink on multiple methods of DNA extraction
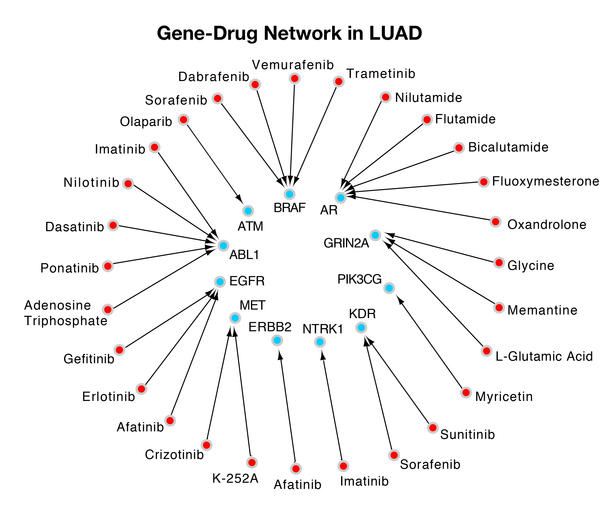
Wang and Gong developed a novel dynamic gene-searching algorithm called Dynamic Gene Search (DyGS) to create a gene panel for each of the 12 cancers with the highest annual incidence and death rate. The 12 gene panels the DyGS algorithm selected used only 3.5% of the original gene mutation pool, while covering every patient sample. About 40% of each gene panel is druggable, which indicates that the DyGS-generated gene panels can be used for early cancer detection as well as therapeutic targets in treatment methods.
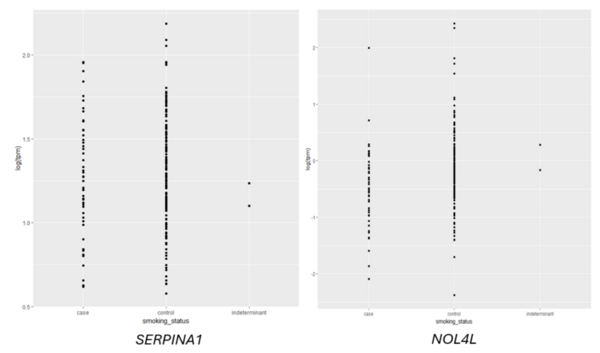
Given an association between nicotine addiction and gene expression, we hypothesized that expression of genes commonly associated with smoking status would have variable expression between smokers and non-smokers. To test whether gene expression varies between smokers and non-smokers, we analyzed two publicly-available datasets that profiled RNA gene expression from brain (nucleus accumbens) and lung tissue taken from patients identified as smokers or non-smokers. We discovered statistically significant differences in expression of dozens of genes between smokers and non-smokers. To test whether gene expression can be used to predict whether a patient is a smoker or non-smoker, we used gene expression as the training data for a logistic regression or random forest classification model. The random forest classifier trained on lung tissue data showed the most robust results, with area under curve (AUC) values consistently between 0.82 and 0.93. Both models trained on nucleus accumbens data had poorer performance, with AUC values consistently between 0.65 and 0.7 when using random forest. These results suggest gene expression can be used to predict smoking status using traditional machine learning models. Additionally, based on our random forest model, we proposed KCNJ3 and TXLNGY as two candidate markers of smoking status. These findings, coupled with other genes identified in this study, present promising avenues for advancing applications related to the genetic foundation of smoking-related characteristics.
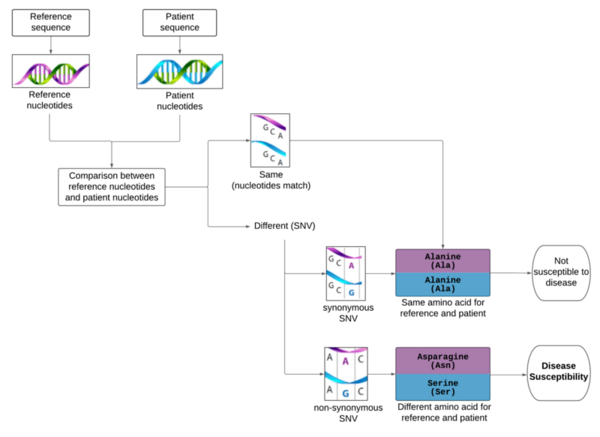
Although the 5-year survival rate for colorectal cancer is below 10%, it increases to greater than 90% if it is diagnosed early. We hypothesized from our research that analyzing non-synonymous single nucleotide variants (SNVs) in a patient's exome sequence would be an indicator for high genetic risk of developing colorectal cancer.
Here the authors sought to better understand glioma, cancer that occurs in the glial cells of the brain with gene expression profile analysis. They considered the expression of complement system genes across the transcriptional and IDH-mutational subtypes of low-grade glioma and glioblastoma. Based on their results of their differential gene expression analysis, they found that outcomes vary across different glioma subtypes, with evidence suggesting that categorization of the transcriptional subtypes could help inform treatment by providing an expectation for treatment responses.
Sequence accessibility is an important factor affecting gene expression. Sequence accessibility or openness impacts the likelihood that a gene is transcribed and translated into a protein and performs functions and manifests traits. There are many potential factors that affect the accessibility of a gene. In this study, our hypothesis was that the content of nucleotides in a genetic sequence predicts its accessibility. Using a machine learning linear regression model, we studied the relationship between nucleotide content and accessibility.
In the United States, there are currently 17.8 million affected by atopic dermatitis (AD), commonly known as eczema. It is characterized by itching and skin inflammation. AD patients are at higher risk for infections, depression, cancer, and suicide. Genetics, environment, and stress are some of the causes of the disease. With the rise of personalized medicine and the acceptance of gene-editing technologies, AD-related variations need to be identified for treatment. Genome-wide association studies (GWAS) have associated the Filaggrin (FLG) gene with AD but have not identified specific problematic single nucleotide polymorphisms (SNPs). This research aimed to refine known SNPs of FLG for gene editing technologies to establish a causal link between specific SNPs and the diseases and to target the polymorphisms. The research utilized R and its Bioconductor packages to refine data from the National Center for Biotechnology Information's (NCBI's) Variation Viewer. The algorithm filtered the dataset by coding regions and conserved domains. The algorithm also removed synonymous variations and treated non-synonymous, frameshift, and nonsense separately. The non-synonymous variations were refined and ordered by the BLOSUM62 substitution matrix. Overall, the analysis removed 96.65% of data, which was redundant or not the focus of the research and ordered the remaining relevant data by impact. The code for the project can also be repurposed as a tool for other diseases. The research can help solve GWAS's imprecise identification challenge. This research is the first step in providing the refined databases required for gene-editing treatment.
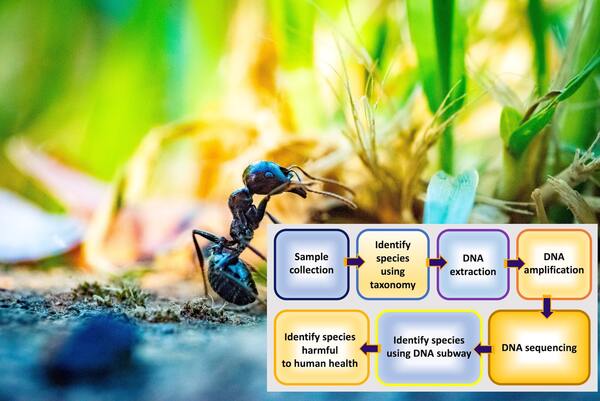
Here the authors used morphological characters and DNA barcoding to identify arthropods found within a residential house. With this method they identified their species and compared them against pests lists provided by the US government. They found that none of their identified species were considered to be pests providing evidence against the misconception that arthropods found at home are harmful to humans. They suggest that these methods could be used at larger scales to better understand and aid in mapping ecosystems.
The sequence of nitrogenous bases that make up the DNA of organisms can contain hidden mathematical sequences. Here the authors used BioPython, a programming tool, to find an organism that displays Gijswijt’s Sequence in its genome. In this manner they found that the common carp best displays Gijswijt’s Sequence in its genome.
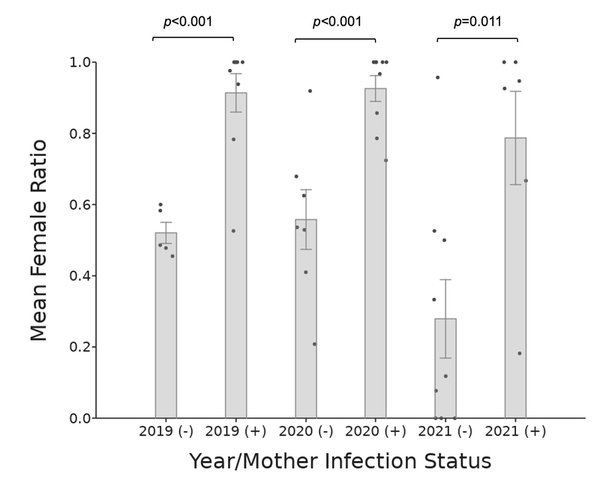
Wolbachia pipientis (Wolbachia) is a maternally inherited endosymbiotic bacterium that infects over 50% of arthropods, including pillbugs, and acts as a reproductive parasite in the host. In the common terrestrial pillbug Armadillidium vulgare (A. vulgare), Wolbachia alters the sex ratio of offspring through a phenomenon called feminization, where genetic males develop into reproductive females. Previous studies have focused on the presence or absence of Wolbachia as a sex ratio distorter in laboratory cultured and natural populations mainly from sites in Europe and Japan. Our three-year study is the first to evaluate the effects of the Wolbachia sex ratio distorter in cultured A. vulgare offspring in North America. We asked whether Wolbachia bacteria feminize A. vulgare isopod male offspring from infected mothers and if this effect can be detected in F1 offspring by comparing the male/female offspring ratios. If so, the F1 offspring ratio should show a higher number of females than males compared to the offspring of uninfected mothers. Over three years, pillbug offspring were cultured from pregnant A. vulgare females and developed into adults. We determined the Wolbachia status of mothers and counted the ratios of male and female F1 progeny to determine feminization effects. In each year sampled, significantly more female offspring were born to Wolbachia-infected mothers than those from uninfected mothers. These ratio differences suggest that the Wolbachia infection status of mothers directly impacts the A. vulgare population through the production of reproductive feminized males, which in turn provides an advantage for further Wolbachia transmission.
_(35622760083).jpg)