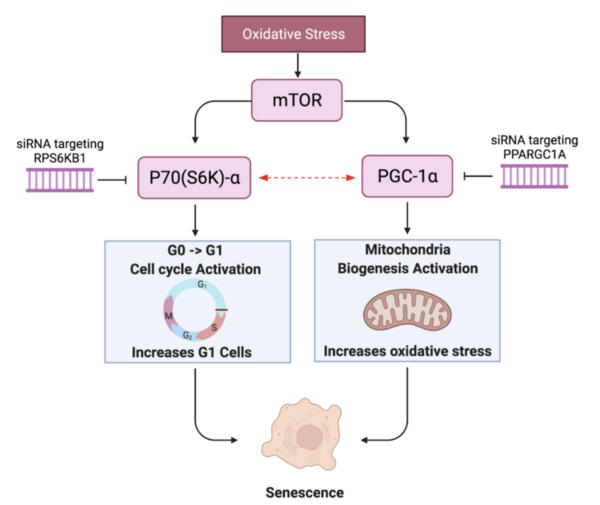
Cellular senescence plays a key role in aging cells and is attributed to a number of disease and pathology. These authors find that genetic editing of both RPS6KB1 and PPARGC1A revitalizes a human skin fibroblast cell line.
Read More...Innovative Treatment for Reducing Senescence and Revitalizing Aging Cells through Gene Silencing
Cellular senescence plays a key role in aging cells and is attributed to a number of disease and pathology. These authors find that genetic editing of both RPS6KB1 and PPARGC1A revitalizes a human skin fibroblast cell line.
Read More...The Impacts of Varying Types of Light on the Growth of Five Arabidopsis Varieties

Arabadopsis, “the fruit fly of plants”, is an easy to grow plant system for genetic manipulation. Here, researchers tested the effects of varied light conditions on plants with specific mutations in the light sensing pathways.
Read More...Development of a Novel Treatment Strategy to Treat Parkinsonian Neurodegeneration by Targeting Both Lewy Body Aggregation and Dopaminergic Neuronal Degradation in a Drosophila melanogaster Model
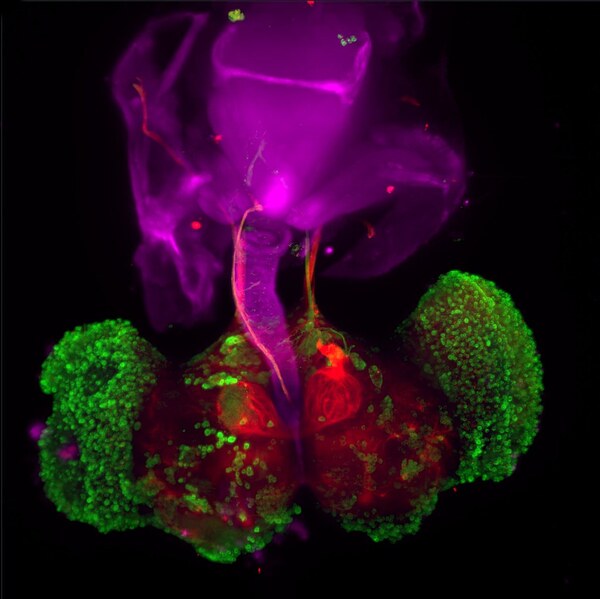
In this article the authors address the complex and life quality-diminishing neurodegenerative disease known as Parkinson's. Although genetic and/or environmental factors contribute to the etiology of the disease, the diagnostic symptoms are the same. By genetically modifying fruit flies to exhibit symptoms of Parkinson's disease, they investigate whether drugs that inhibit mitochondrial calcium uptake or activate the lysosomal degradation of proteins could improve the symptoms of Parkinson's these flies exhibit. The authors report the most promising outcome to be that when both types of drugs were used together. Their data provides encouraging evidence to support further investigation of the utility of such drugs in the treatment of human Parkinson's patients.
Read More...Impact of daf-25 and daf-11 Mutations on Olfactory Function in C. elegans
Cilia are little hair-like protrusions on many cells in the human body, including those lining the trachea where they play a role in clearing our respiratory tract of mucous and other irritants. Genetic mutations that impair ciliary function have serious consequences on our well-being making it important to understand how ciliary function is regulated. By using a simple organism, such as the worm C. elegans that use cilia to move, the authors explore the effect of certain genetic mutations on the cilia of the worms by measuring their ability to move towards or away from certain odorants.
Read More...Male Feminization of the Common Pillbug Armadillidium vulgare by Wolbachia bacteria
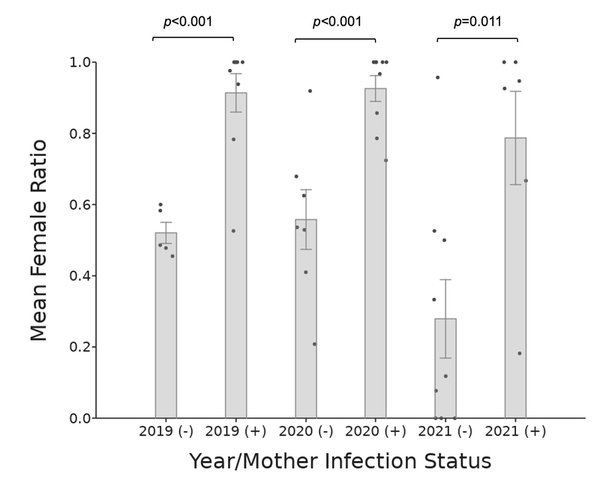
Wolbachia pipientis (Wolbachia) is a maternally inherited endosymbiotic bacterium that infects over 50% of arthropods, including pillbugs, and acts as a reproductive parasite in the host. In the common terrestrial pillbug Armadillidium vulgare (A. vulgare), Wolbachia alters the sex ratio of offspring through a phenomenon called feminization, where genetic males develop into reproductive females. Previous studies have focused on the presence or absence of Wolbachia as a sex ratio distorter in laboratory cultured and natural populations mainly from sites in Europe and Japan. Our three-year study is the first to evaluate the effects of the Wolbachia sex ratio distorter in cultured A. vulgare offspring in North America. We asked whether Wolbachia bacteria feminize A. vulgare isopod male offspring from infected mothers and if this effect can be detected in F1 offspring by comparing the male/female offspring ratios. If so, the F1 offspring ratio should show a higher number of females than males compared to the offspring of uninfected mothers. Over three years, pillbug offspring were cultured from pregnant A. vulgare females and developed into adults. We determined the Wolbachia status of mothers and counted the ratios of male and female F1 progeny to determine feminization effects. In each year sampled, significantly more female offspring were born to Wolbachia-infected mothers than those from uninfected mothers. These ratio differences suggest that the Wolbachia infection status of mothers directly impacts the A. vulgare population through the production of reproductive feminized males, which in turn provides an advantage for further Wolbachia transmission.
Read More...Using explainable artificial intelligence to identify patient-specific breast cancer subtypes
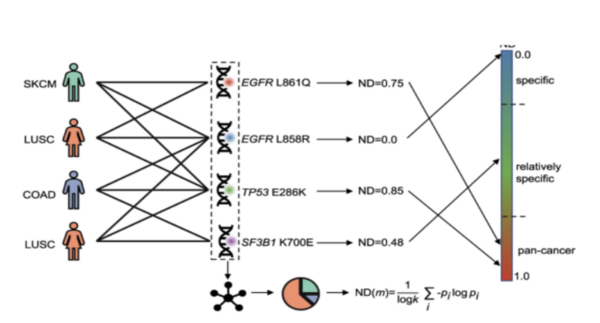
Breast cancer is the most common cancer in women, with approximately 300,000 diagnosed with breast cancer in 2023. It ranks second in cancer-related deaths for women, after lung cancer with nearly 50,000 deaths. Scientists have identified important genetic mutations in genes like BRCA1 and BRCA2 that lead to the development of breast cancer, but previous studies were limited as they focused on specific populations. To overcome limitations, diverse populations and powerful statistical methods like genome-wide association studies and whole-genome sequencing are needed. Explainable artificial intelligence (XAI) can be used in oncology and breast cancer research to overcome these limitations of specificity as it can analyze datasets of diagnosed patients by providing interpretable explanations for identified patterns and predictions. This project aims to achieve technological and medicinal goals by using advanced algorithms to identify breast cancer subtypes for faster diagnoses. Multiple methods were utilized to develop an efficient algorithm. We hypothesized that an XAI approach would be best as it can assign scores to genes, specifically with a 90% success rate. To test that, we ran multiple trials utilizing XAI methods through the identification of class-specific and patient-specific key genes. We found that the study demonstrated a pipeline that combines multiple XAI techniques to identify potential biomarker genes for breast cancer with a 95% success rate.
Read More...Can the nucleotide content of a DNA sequence predict the sequence accessibility?

Sequence accessibility is an important factor affecting gene expression. Sequence accessibility or openness impacts the likelihood that a gene is transcribed and translated into a protein and performs functions and manifests traits. There are many potential factors that affect the accessibility of a gene. In this study, our hypothesis was that the content of nucleotides in a genetic sequence predicts its accessibility. Using a machine learning linear regression model, we studied the relationship between nucleotide content and accessibility.
Read More...Expression of Anti-Neurodegeneration Genes in Mutant Caenorhabditis elegans Using CRISPR-Cas9 Improves Behavior Associated With Alzheimer’s Disease
Alzheimer's disease is one of the leading causes of death in the United States and is characterized by neurodegeneration. Mishra et al. wanted to understand the role of two transport proteins, LRP1 and AQP4, in the neurodegeneration of Alzheimer's disease. They used a model organism for Alzheimer's disease, the nematode C. elegans, and genetic engineering to look at whether they would see a decrease in neurodegeneration if they increased the amount of these two transport proteins. They found that the best improvements were caused by increased expression of both transport proteins, with smaller improvements when just one of the proteins is overly expressed. Their work has important implications for how we understand neurodegeneration in Alzheimer's disease and what we can do to slow or prevent the progression of the disease.
Read More...Predicting smoking status based on RNA sequencing data
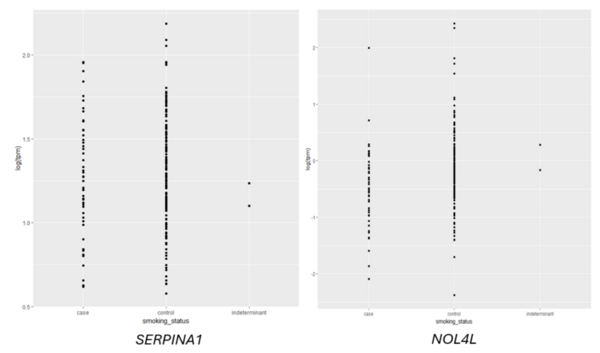
Given an association between nicotine addiction and gene expression, we hypothesized that expression of genes commonly associated with smoking status would have variable expression between smokers and non-smokers. To test whether gene expression varies between smokers and non-smokers, we analyzed two publicly-available datasets that profiled RNA gene expression from brain (nucleus accumbens) and lung tissue taken from patients identified as smokers or non-smokers. We discovered statistically significant differences in expression of dozens of genes between smokers and non-smokers. To test whether gene expression can be used to predict whether a patient is a smoker or non-smoker, we used gene expression as the training data for a logistic regression or random forest classification model. The random forest classifier trained on lung tissue data showed the most robust results, with area under curve (AUC) values consistently between 0.82 and 0.93. Both models trained on nucleus accumbens data had poorer performance, with AUC values consistently between 0.65 and 0.7 when using random forest. These results suggest gene expression can be used to predict smoking status using traditional machine learning models. Additionally, based on our random forest model, we proposed KCNJ3 and TXLNGY as two candidate markers of smoking status. These findings, coupled with other genes identified in this study, present promising avenues for advancing applications related to the genetic foundation of smoking-related characteristics.
Read More...Investigating the inhibition of catabolic enzymes for implications in cardiovascular diseases and diabetes
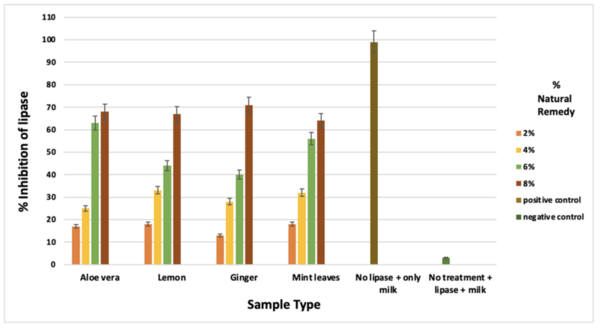
Enzymes that metabolize carbohydrates and lipids play a key role in our health, including global health challenges like cardiovascular diseases and diabetes. To learn more about these important enzymes, Gandhi and Gandhi test whether various natural substances (ginger, Aloe vera, lemon, and mint leaves) affect the activity of α-amylase and lipase enzymes.
Read More...