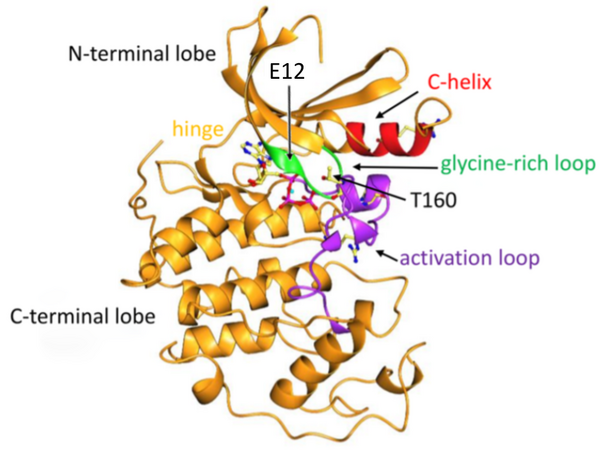
The authors examined the impact of mutations to CDK2 on protein expression.
Read More...Investigating the effects of mutations of amino acids on the protein expression of CDK2 cancer gene
Effects on Learning and Memory of a Mutation in Dα7: A D. melanogaster Homolog of Alzheimer's Related Gene for nAChR α7

Alzheimer's disease (AD) involves the reduction of cholinergic activity due to a decrease in neuronal levels of nAChR α7. In this work, Sanyal and Cuellar-Ortiz explore the role of the nAChR α7 in learning and memory retention, using Drosophila melanogaster as a model organism. The performance of mutant flies (PΔEY6) was analyzed in locomotive and olfactory-memory retention tests in comparison to wild type (WT) flies and an Alzheimer's disease model Arc-42 (Aβ-42). Their results suggest that the lack of the D. melanogaster-nAChR causes learning, memory, and locomotion impairments, similar to those observed in Alzheimer's models Arc-42.
Read More...Activated NF-κB Pathway in an Irf6-Deficient Mouse Model for Van der Woude Syndrome
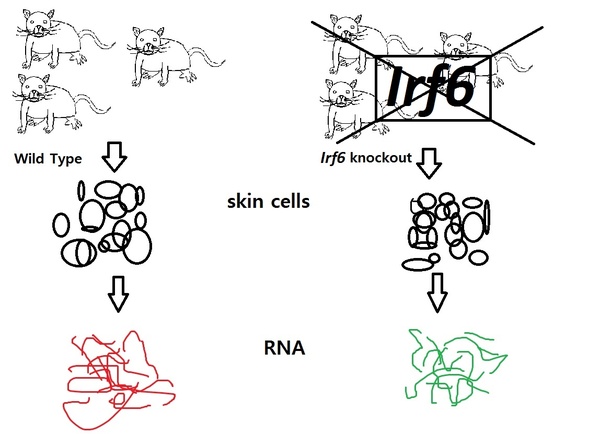
Van der Woude syndrome is a common birth defect caused by mutations in the gene Irf6. In this project, students used microarray expression analysis from wild-type and Irf6-deficient mice in order to identify gene networks or pathways differentially regulated due to the Irf6 mutation. They found NF-κB pathway to be activated in deficient mice.
Read More...Effects of urban traffic noise on the early growth and transcription of Arabidopsis thaliana
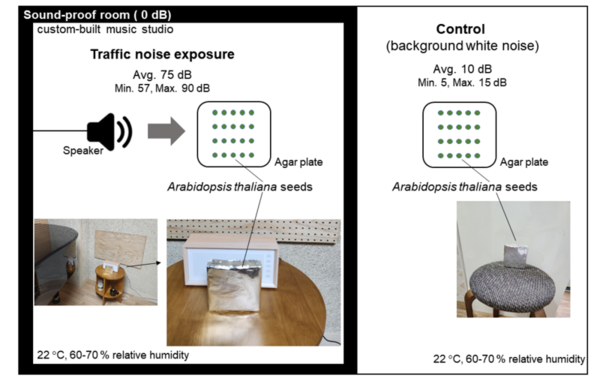
This article explores the largely unstudied impact of noise pollution on plant life. By exposing Arabidopsis thaliana seedlings to urban traffic noise, the study found a significant increase in seedling growth, alongside substantial changes in gene expression. This research reveals critical insights into how noise pollution affects plant physiology and contributes to a broader understanding of its ecological impacts, helping to guide future efforts in ecosystem conservation.
Read More...Predicting smoking status based on RNA sequencing data
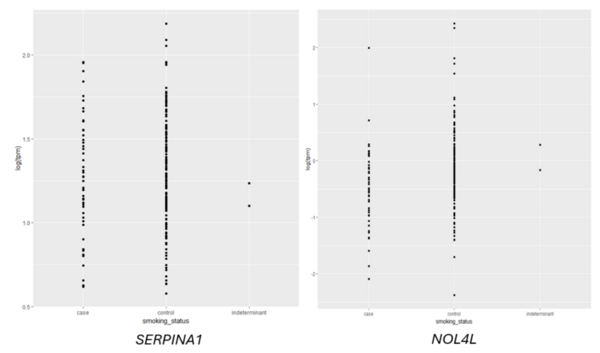
Given an association between nicotine addiction and gene expression, we hypothesized that expression of genes commonly associated with smoking status would have variable expression between smokers and non-smokers. To test whether gene expression varies between smokers and non-smokers, we analyzed two publicly-available datasets that profiled RNA gene expression from brain (nucleus accumbens) and lung tissue taken from patients identified as smokers or non-smokers. We discovered statistically significant differences in expression of dozens of genes between smokers and non-smokers. To test whether gene expression can be used to predict whether a patient is a smoker or non-smoker, we used gene expression as the training data for a logistic regression or random forest classification model. The random forest classifier trained on lung tissue data showed the most robust results, with area under curve (AUC) values consistently between 0.82 and 0.93. Both models trained on nucleus accumbens data had poorer performance, with AUC values consistently between 0.65 and 0.7 when using random forest. These results suggest gene expression can be used to predict smoking status using traditional machine learning models. Additionally, based on our random forest model, we proposed KCNJ3 and TXLNGY as two candidate markers of smoking status. These findings, coupled with other genes identified in this study, present promising avenues for advancing applications related to the genetic foundation of smoking-related characteristics.
Read More...Can the nucleotide content of a DNA sequence predict the sequence accessibility?

Sequence accessibility is an important factor affecting gene expression. Sequence accessibility or openness impacts the likelihood that a gene is transcribed and translated into a protein and performs functions and manifests traits. There are many potential factors that affect the accessibility of a gene. In this study, our hypothesis was that the content of nucleotides in a genetic sequence predicts its accessibility. Using a machine learning linear regression model, we studied the relationship between nucleotide content and accessibility.
Read More...Disruptions in protein-protein interactions between HTT, PRPF40B, and MECP2 are involved in Lopes-Maciel-Rodan syndrome
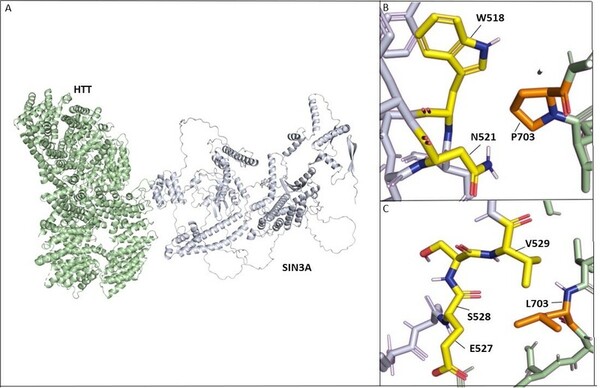
In an extensive study of gene mutations, and their resulting effect on protein-protein interactions, Desai and Stork found that HTT-PRPF40B-MECP2 interactions are weakened with progression of Lopes-Maciel-Rodan syndrome.
Read More...Expressional correlations between SERPINA6 and pancreatic ductal adenocarcinoma-linked genes
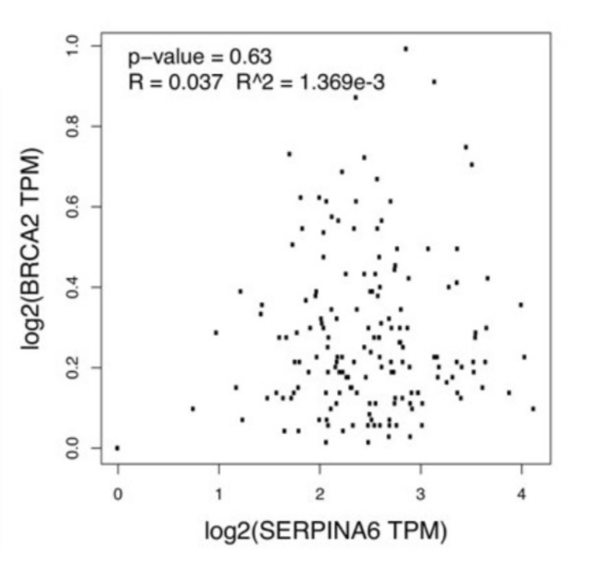
Pancreatic ductal adenocarcinoma (PDAC) is the most common form of pancreatic cancer, with early diagnosis and treatment challenges. When any of the genes KRAS, SMAD4, TP53, and BRCA2 are heavily mutated, they correlate with PDAC progression. Cellular stress, partly regulated by the gene SERPINA6, also correlates with PDAC progression. When SERPINA6 is highly expressed, corticosteroid-binding globulin inhibits the effect of the stress hormone cortisol. In this study, the authors explored whether there is an inverse correlation between the expression of SERPINA6 and PDAC-linked genes.
Read More...Evolution of Neuroplastin-65
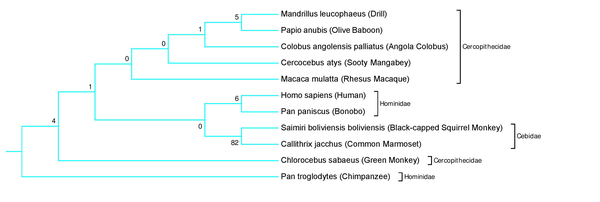
Human intelligence is correlated with variation in the protein neuroplastin-65, which is encoded by the NPTN gene. The authors examine the evolution of this gene across different animal species.
Read More...Upregulation of the Ribosomal Pathway as a Potential Blood-Based Genetic Biomarker for Comorbid Major Depressive Disorder (MDD) and PTSD
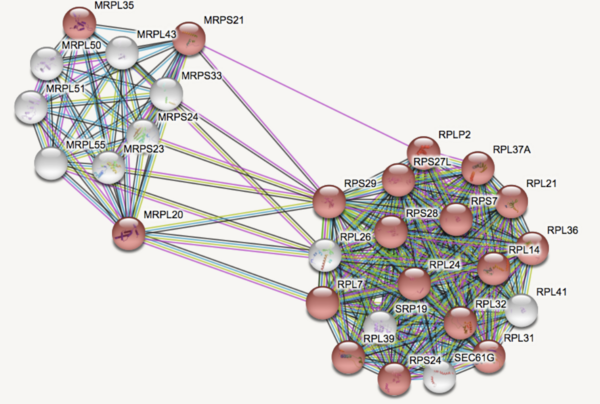
Major Depressive Disorder (MDD), and Post-Traumatic Stress Disorder (PTSD) are two of the fastest growing comorbid diseases in the world. Using publicly available datasets from the National Institute for Biotechnology Information (NCBI), Ravi and Lee conducted a differential gene expression analysis using 184 blood samples from either control individuals or individuals with comorbid MDD and PTSD. As a result, the authors identified 253 highly differentially-expressed genes, with enrichment for proteins in the gene ontology group 'Ribosomal Pathway'. These genes may be used as blood-based biomarkers for susceptibility to MDD or PTSD, and to tailor treatments within a personalized medicine regime.
Read More...