Sequence accessibility is an important factor affecting gene expression. Sequence accessibility or openness impacts the likelihood that a gene is transcribed and translated into a protein and performs functions and manifests traits. There are many potential factors that affect the accessibility of a gene. In this study, our hypothesis was that the content of nucleotides in a genetic sequence predicts its accessibility. Using a machine learning linear regression model, we studied the relationship between nucleotide content and accessibility.
Cilia are little hair-like protrusions on many cells in the human body, including those lining the trachea where they play a role in clearing our respiratory tract of mucous and other irritants. Genetic mutations that impair ciliary function have serious consequences on our well-being making it important to understand how ciliary function is regulated. By using a simple organism, such as the worm C. elegans that use cilia to move, the authors explore the effect of certain genetic mutations on the cilia of the worms by measuring their ability to move towards or away from certain odorants.
While the genetic basis of hip fracture risk has been studied extensively in adults, it is not known whether parental history of bone fractures affects their children's fracture risk. In this article, the authors investigated whether a parental history of bone fractures influences the rate of fractures in their children. They found that adolescent children whose parents had a more extensive history of fractures were more likely to have a history of fractures themselves, suggesting that parents' medical histories may be an important consideration in future pediatric health research.
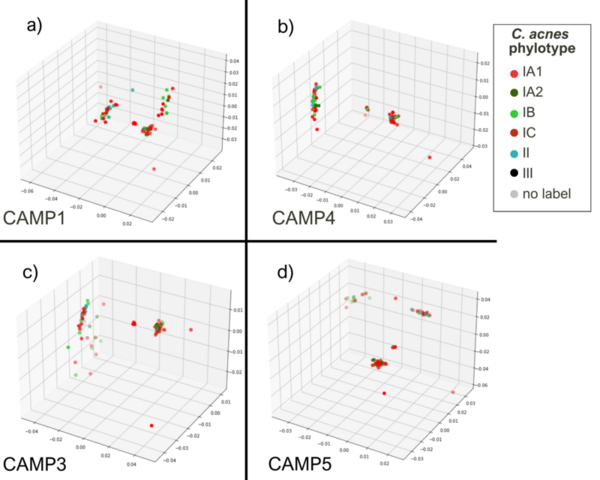
Cutibacterium acnes is a bacterium believed to play an important role in the pathogenesis of common skin diseases such as acne vulgaris. Currently, acne is known to be associated with strains from the type IA1 and IC clades of C. acnes, while those from the type IA2, IB, II, and III phylogroups are associated with skin health. This is the first study to explore the sequence space of individual gene products of different C. acnes phylogroups. Our analysis compared the sequence space topology of virulence factors to proteins with unknown functions and housekeeping proteins. We hypothesized that sequence space features of virulence factors are different from housekeeping protein features, which potentially provides an avenue to deduce unknown proteins’ functions. This proposition should be confirmed based on further experimental outcomes. A notable similarity in the sequence spaces’ topological features of previously known as housekeeping proteins encoded by recA and guaA genes to ‘putative virulence’ genes camp2 and tly was observed. Our research suggests further investigation of recA and guaA’s potential virulence properties to better understand acne pathogenesis and develop more targeted acne treatments.
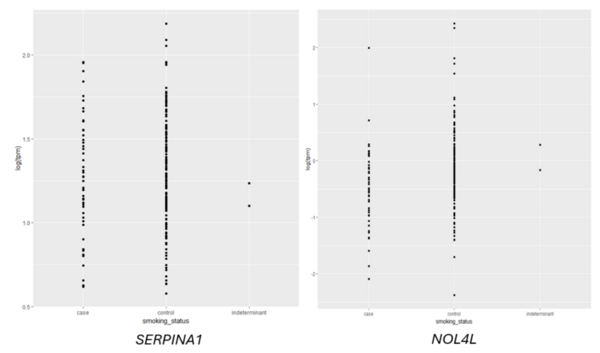
Given an association between nicotine addiction and gene expression, we hypothesized that expression of genes commonly associated with smoking status would have variable expression between smokers and non-smokers. To test whether gene expression varies between smokers and non-smokers, we analyzed two publicly-available datasets that profiled RNA gene expression from brain (nucleus accumbens) and lung tissue taken from patients identified as smokers or non-smokers. We discovered statistically significant differences in expression of dozens of genes between smokers and non-smokers. To test whether gene expression can be used to predict whether a patient is a smoker or non-smoker, we used gene expression as the training data for a logistic regression or random forest classification model. The random forest classifier trained on lung tissue data showed the most robust results, with area under curve (AUC) values consistently between 0.82 and 0.93. Both models trained on nucleus accumbens data had poorer performance, with AUC values consistently between 0.65 and 0.7 when using random forest. These results suggest gene expression can be used to predict smoking status using traditional machine learning models. Additionally, based on our random forest model, we proposed KCNJ3 and TXLNGY as two candidate markers of smoking status. These findings, coupled with other genes identified in this study, present promising avenues for advancing applications related to the genetic foundation of smoking-related characteristics.
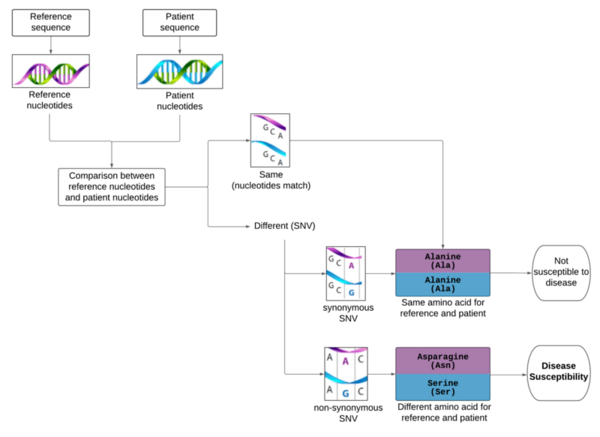
Although the 5-year survival rate for colorectal cancer is below 10%, it increases to greater than 90% if it is diagnosed early. We hypothesized from our research that analyzing non-synonymous single nucleotide variants (SNVs) in a patient's exome sequence would be an indicator for high genetic risk of developing colorectal cancer.
Here the authors sought to better understand glioma, cancer that occurs in the glial cells of the brain with gene expression profile analysis. They considered the expression of complement system genes across the transcriptional and IDH-mutational subtypes of low-grade glioma and glioblastoma. Based on their results of their differential gene expression analysis, they found that outcomes vary across different glioma subtypes, with evidence suggesting that categorization of the transcriptional subtypes could help inform treatment by providing an expectation for treatment responses.
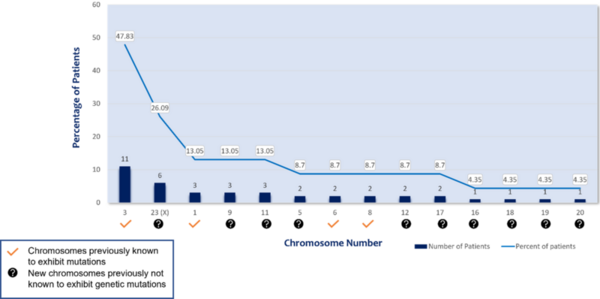
Uveal melanoma (UM) is a rare subtype of melanoma but the most frequent primary cancer of the eye in adults. The goal of this study was to research the genetic causes of UM through a comprehensive frequency analysis of base-pair mismatches in patient genomes. Results showed a total of 130 genetic mutations, including seven recurrent mutations, with most mutations occurring in chromosomes 3 and X. Recurrent mutations varied from 8.7% to 17.39% occurrence in the UM patient sample, with all mutations identified as missense. These findings suggest that UM is a recessive heterogeneous disease with selective homozygous mutations. Notably, this study has potential wider significance because the seven genes targeted by recurrent mutations are also involved in other cancers.
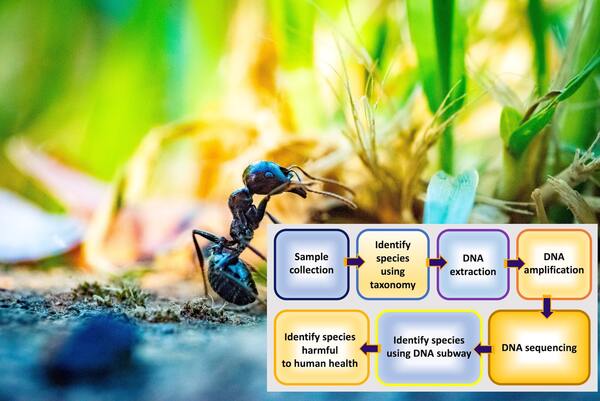
Here the authors used morphological characters and DNA barcoding to identify arthropods found within a residential house. With this method they identified their species and compared them against pests lists provided by the US government. They found that none of their identified species were considered to be pests providing evidence against the misconception that arthropods found at home are harmful to humans. They suggest that these methods could be used at larger scales to better understand and aid in mapping ecosystems.