In silico design of novel acetylcholinesterase inhibitors as potential therapeutics for Alzheimer's disease
(1) Northfield Mount Hermon School
https://doi.org/10.59720/25-140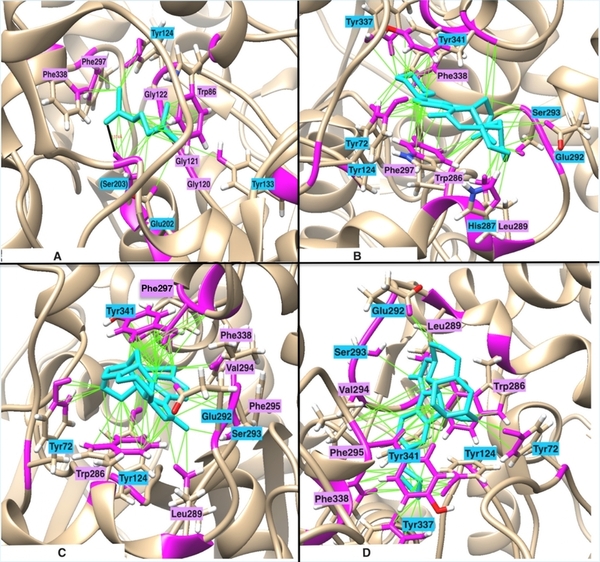
Scientific literature suggests that the elevated activity of acetylcholinesterase (AChE) in breaking down the neurotransmitter acetylcholine (ACh) is the main contributing factor leading to cognitive decline and neurodegenerative diseases, including Alzheimer’s disease (AD). However, due to the poor bioavailability of existing AChE inhibitors, the demand for new drugs to be developed persists. In this paper, we designed novel, non-toxic, competitive inhibitors targeting AChE from structures of 72 drugs targeting AChE, using computational techniques including molecular docking, chemical analysis, molecular modification, and molecular dynamics (MD) simulation. Since AChE's binding site contains active residues that possess polar aromatic side chains, we hypothesized that adding aromatic R-groups to ligands would promote hydrogen bonding and pi-pi interactions, thereby increasing the thermodynamic favorability of AChE-ligand binding. In accordance with our hypothesis, drugs with high logP (high hydrophobicity) and molar refractivity (high polarizability) had slightly stronger predicted binding energies. In fact, maintaining high logP and molar refractivity, ligands 17 and 18 exhibited the highest predicted binding energies. Moreover, we deemed them as non-toxic toward human cell culture, as they passed the Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) tests. However, when compared to ligand 17 simple chloroethyl side chain, our MD results indicated that bulky or cyclic R-groups in ligand 18 led to less stable binding conformations due to induced steric hindrance in the AChE active site. With these results highlighting the promise of AChE inhibitors, in vivo experiments are required to validate the inhibitory efficacy and cytotoxicity of ligand 17 as a potential-enhanced AD therapeutic.
This article has been tagged with: