Disruptions in protein-protein interactions between HTT, PRPF40B, and MECP2 are involved in Lopes-Maciel-Rodan syndrome
(1) Juanita High School , (2) Harvard University
https://doi.org/10.59720/21-180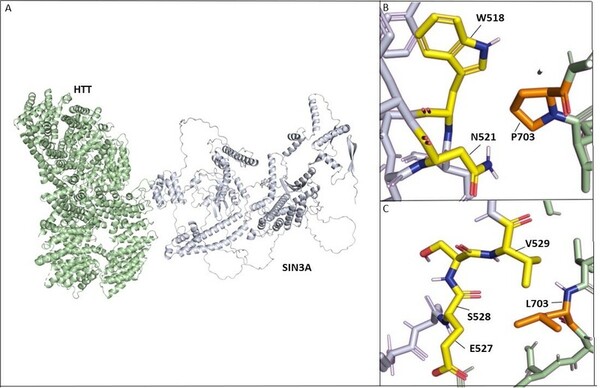
Lopes-Maciel-Rodan syndrome (LOMARS) is a rare and serious neurodevelopmental disorder caused by two compound heterozygous missense mutations in the Huntingtin gene (HTT). LOMARS is related to Rett syndrome, which is primarily caused by mutations in X-linked Methyl-CpG-Binding Protein 2 (MECP2), since it results in Rett-like neurological phenotypes and manifestations. While HTT and MECP2 proteins are not known to directly bind to each other, it is plausible that other protein(s) enable and enhance this protein-protein interaction (PPI). This study reports the involvement of pre-mRNA Processing Factor 40 Homolog B (PRPF40B) in mediating the PPI between HTT and MECP2. By using coevolution data, we found that PRPF40B interacts with both HTT and MECP2 through its group II WW domain, a modular protein domain that mediates protein-protein interactions. We also revealed that multiple missense and splice region variants in PRPF40B result in LOMARS and Rett-like phenotypes, suggesting that weakened interactions between mutant PRPF40B and wild-type MECP2 are likely to be associated with the phenotypes. Furthermore, upon docking wild-type and mutant models of HTT with PRPF40B, we observed that the LOMARS-associated mutations significantly weaken HTT-PRPF40B interactions. We also performed similar docking experiments on SIN3A and PRPF40A, which is a protein that is highly related in structure and function to PRPF40B and observed that the mutations also weaken their interactions with HTT. Overall, this study demonstrates the significant role of HTT-PRPF40B-MECP2 interactions in the development and progression of LOMARS and suggests the involvement of similar PPI disruption in Rett syndrome.
This article has been tagged with: