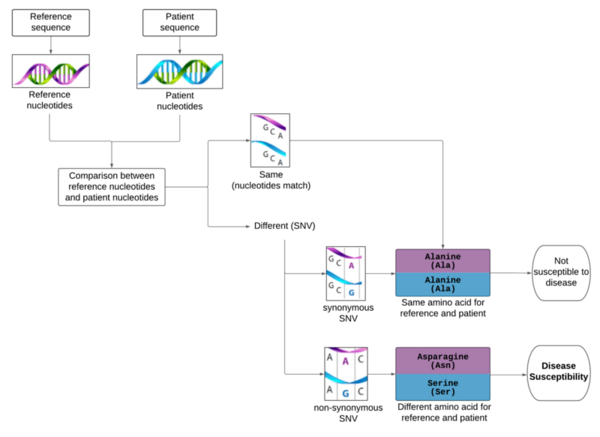
Cystic fibrosis is a genetic disease caused by mutations in the CFTR gene. In this paper, the authors attempt to identify variations in stretches of up to 8 nucleotides in the protein-coding portions of the CFTR gene that are associated with disease development. This would allow screening of newborns or even fetuses in utero to determine the likelihood they develop cystic fibrosis.
In this study, the authors use bioinformatic approaches to characterize the mirror neurons, which are active when performing and seeing certain actions. They also investigated whether mirror neuron impairment was connected to neural degenerative diseases and psychiatric disorders.
Recent declines in the brook trout population of the Lake Champlain Basin have made the genetic screening of this and other trout species of utmost importance. In this study, the authors collected and analyzed 21 DNA samples from Lake Champlain Basin trout populations and performed a phylogenetic analysis on these samples using the cytochrome b gene. The findings presented in this study may influence future habitat decisions in this region.
Here the authors investigated a combination therapy to target the Kirsten rat sarcoma viral oncogene homolog mutation in lung cancer, by analyzing publicly available data. Their findings indicate that the combination therapy of CA170 and Kvax enhances helper T cell function and improves cytotoxic T lymphocyte infiltration, while Kvax alone drives plasma and memory B cell proliferation.
Here, the authors investigated the integration of large language models (LLMs) with drug target affinity predictors (DTAPs) to improve drug repurposing, demonstrating a significant increase in prediction accuracy, particularly with GPT-4, for psychotropic drugs and the sigma-1 receptor. This novel approach offers to potentially accelerate and reduce the cost of drug discovery by efficiently identifying new therapeutic uses for existing drugs.
Here, recognizing the difficulty associated with tracking the progression of dementia, the authors used machine learning models to predict between the presence of cognitive normalcy, mild cognitive impairment, and Alzheimer's Disease, based on blood DNA methylation levels, sex, and age. With four machine learning models and two dataset dimensionality reduction methods they achieved an accuracy of 53.33%.
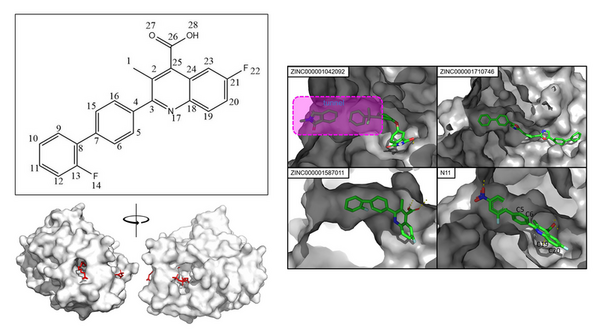
Here, seeking to address the growing threat of multidrug-resistant bacteria (MDR). the authors used in silico virtual screening to target MDR Pseudomonas aeruginosa. They considered a key protein in its biosynthesis and virtually screened 20,000 candidates and 30 derivatives of brequinar. In the end, they identified a possible candidate with the highest degree of potential to inhibit the pathogen's lipid A synthesis.
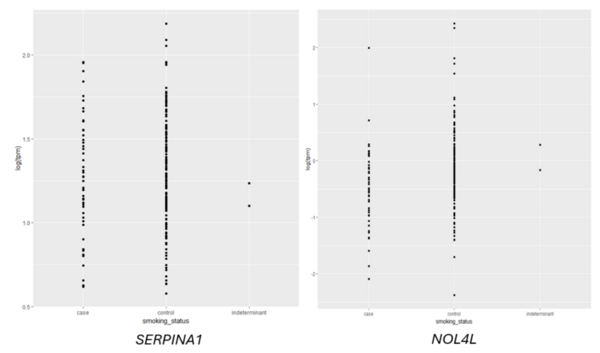
Given an association between nicotine addiction and gene expression, we hypothesized that expression of genes commonly associated with smoking status would have variable expression between smokers and non-smokers. To test whether gene expression varies between smokers and non-smokers, we analyzed two publicly-available datasets that profiled RNA gene expression from brain (nucleus accumbens) and lung tissue taken from patients identified as smokers or non-smokers. We discovered statistically significant differences in expression of dozens of genes between smokers and non-smokers. To test whether gene expression can be used to predict whether a patient is a smoker or non-smoker, we used gene expression as the training data for a logistic regression or random forest classification model. The random forest classifier trained on lung tissue data showed the most robust results, with area under curve (AUC) values consistently between 0.82 and 0.93. Both models trained on nucleus accumbens data had poorer performance, with AUC values consistently between 0.65 and 0.7 when using random forest. These results suggest gene expression can be used to predict smoking status using traditional machine learning models. Additionally, based on our random forest model, we proposed KCNJ3 and TXLNGY as two candidate markers of smoking status. These findings, coupled with other genes identified in this study, present promising avenues for advancing applications related to the genetic foundation of smoking-related characteristics.
Although the 5-year survival rate for colorectal cancer is below 10%, it increases to greater than 90% if it is diagnosed early. We hypothesized from our research that analyzing non-synonymous single nucleotide variants (SNVs) in a patient's exome sequence would be an indicator for high genetic risk of developing colorectal cancer.
Plant diseases can cause up to 50% crop yield loss for the popular tomato plant. A mobile device-based method to identify diseases from photos of symptomatic leaves via computer vision can be more effective due to its convenience and accessibility. To enable a practical mobile solution, a “shallow” convolutional neural networks (CNNs) with few layers, and thus low computational requirement but with high accuracy similar to the deep CNNs is needed. In this work, we explored if such a model was possible.