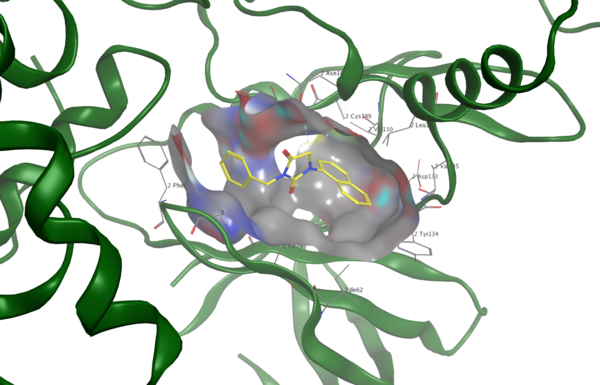
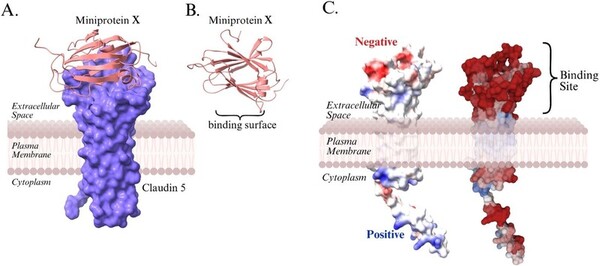
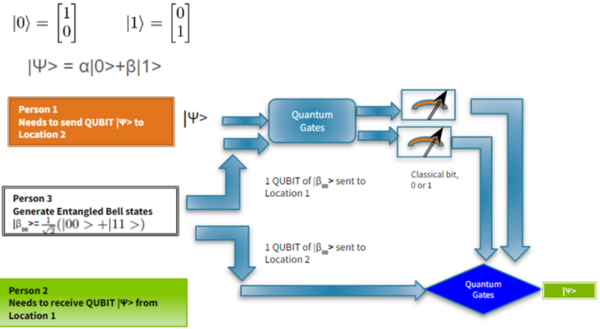
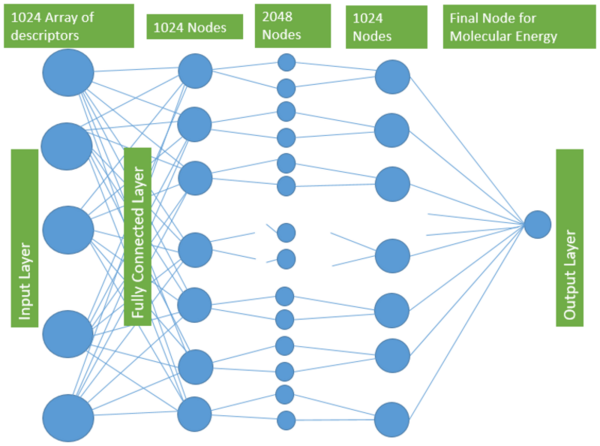
With molecular energy being an integral element to the study of molecules and molecular interactions, computational methods to determine molecular energy are used for the preservation of time and resources. However, these computational methods have high demand for computer resources, limiting their widespread feasibility. The authors of this study employed machine learning to address this disadvantage, utilizing neural networks trained on different representations of molecules to predict molecular properties without the requirement of computationally-intensive processing. In their findings, the authors determined the Feedforward Neural Network, trained by two separate models, as capable of predicting molecular energy with limited prediction error.
Read More...


{kind=link}