The sequence of nitrogenous bases that make up the DNA of organisms can contain hidden mathematical sequences. Here the authors used BioPython, a programming tool, to find an organism that displays Gijswijt’s Sequence in its genome. In this manner they found that the common carp best displays Gijswijt’s Sequence in its genome.
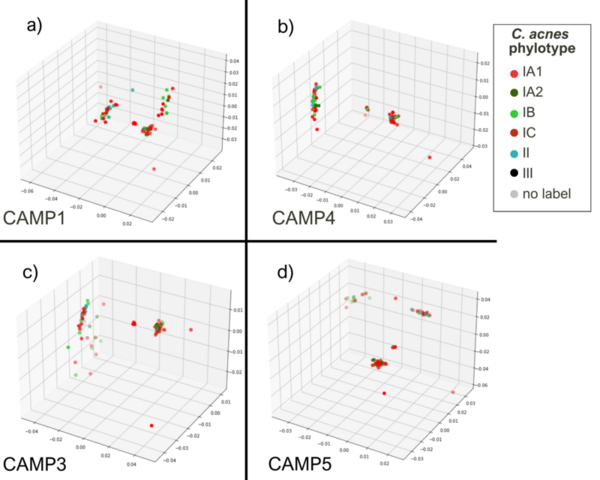
Cutibacterium acnes is a bacterium believed to play an important role in the pathogenesis of common skin diseases such as acne vulgaris. Currently, acne is known to be associated with strains from the type IA1 and IC clades of C. acnes, while those from the type IA2, IB, II, and III phylogroups are associated with skin health. This is the first study to explore the sequence space of individual gene products of different C. acnes phylogroups. Our analysis compared the sequence space topology of virulence factors to proteins with unknown functions and housekeeping proteins. We hypothesized that sequence space features of virulence factors are different from housekeeping protein features, which potentially provides an avenue to deduce unknown proteins’ functions. This proposition should be confirmed based on further experimental outcomes. A notable similarity in the sequence spaces’ topological features of previously known as housekeeping proteins encoded by recA and guaA genes to ‘putative virulence’ genes camp2 and tly was observed. Our research suggests further investigation of recA and guaA’s potential virulence properties to better understand acne pathogenesis and develop more targeted acne treatments.
In the United States, there are currently 17.8 million affected by atopic dermatitis (AD), commonly known as eczema. It is characterized by itching and skin inflammation. AD patients are at higher risk for infections, depression, cancer, and suicide. Genetics, environment, and stress are some of the causes of the disease. With the rise of personalized medicine and the acceptance of gene-editing technologies, AD-related variations need to be identified for treatment. Genome-wide association studies (GWAS) have associated the Filaggrin (FLG) gene with AD but have not identified specific problematic single nucleotide polymorphisms (SNPs). This research aimed to refine known SNPs of FLG for gene editing technologies to establish a causal link between specific SNPs and the diseases and to target the polymorphisms. The research utilized R and its Bioconductor packages to refine data from the National Center for Biotechnology Information's (NCBI's) Variation Viewer. The algorithm filtered the dataset by coding regions and conserved domains. The algorithm also removed synonymous variations and treated non-synonymous, frameshift, and nonsense separately. The non-synonymous variations were refined and ordered by the BLOSUM62 substitution matrix. Overall, the analysis removed 96.65% of data, which was redundant or not the focus of the research and ordered the remaining relevant data by impact. The code for the project can also be repurposed as a tool for other diseases. The research can help solve GWAS's imprecise identification challenge. This research is the first step in providing the refined databases required for gene-editing treatment.
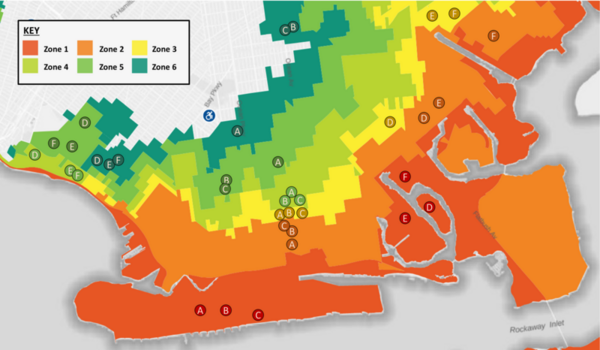
With climate change and rising sea levels, south Brooklyn is exposed to massive flooding and intense precipitation. Previous research discovered that flooding shifts plant species distribution, decreases soil pH, and increases salt concentration, nitrogen, phosphorus, and potassium levels. The authors predicted a decreasing trend from Zone 1 to 6: high-pH, high-salt, and high-nutrients in more flood-prone areas to low-pH, low-salt, and low-nutrient in less flood-prone regions. They performed DNA barcoding to identify plant species inhabiting flood zones with expectations of decreasing salt tolerance and moisture uptake by plants' soil from Zones 1-6. Furthermore, they predicted an increase in invasive species, ultimately resulting in a decrease in biodiversity. After barcoding, they researched existing information regarding invasiveness, ideal soil, pH tolerance, and salt tolerance. They performed soil analyses to identify pH, nitrogen (N), phosphorus (P), and potassium (K) levels. For N and P levels, we discovered a general decreasing trend from Zone 1 to 6 with low and moderate statistical significance respectively. Previous studies found that soil moisture can increase N and P uptake, helping plants adopt efficient resource-use strategies and reduce water stress from flooding. Although characteristics of plants were distributed throughout all zones, demonstrating overall diversity, the soil analyses hinted at the possibility of a rising trend of plants adapting to the increase in flooding. Future expansive research is needed to comprehensively map these trends. Ultimately, investigating trends between flood zones and the prevalence of different species will assist in guiding solutions to weathering climate change and protecting biodiversity in Brooklyn.
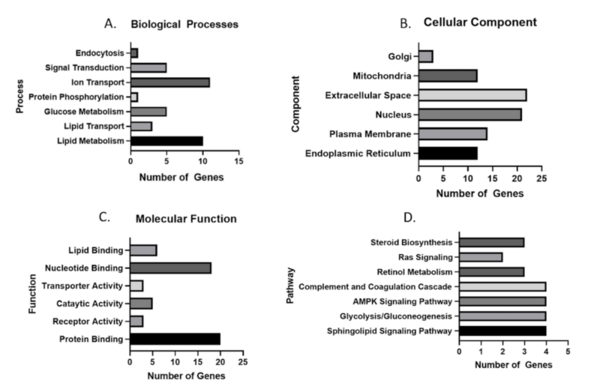
In this study, the authors analyze gene expression datasets to determine if there is a core set of genes dysregulated during nonalcoholic steatohepatitis.
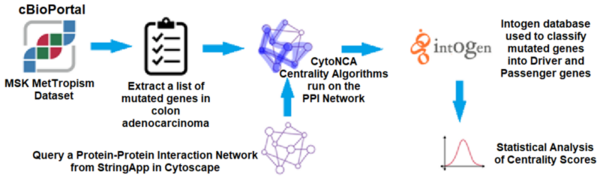
In this article the authors created an interaction map of proteins involved in colorectal cancer to look for driver vs. non-driver genes. That is they wanted to see if they could determine what genes are more likely to drive the development and progression in colorectal cancer and which are present in altered states but not necessarily driving disease progression.
We know relatively little about how vegan diets and non-vegan diets compare when it comes to the gut microbiome. Gollamudi and Gollamudi tackle this challenge by investigating how changes in a participant's diet affected the diversity of their intestinal microbiome.