Identification of a core set of model agnostic mRNA associated with nonalcoholic steatohepatitis (NASH)
(1) John F. Kennedy High School, Bellmore, New York
https://doi.org/10.59720/21-262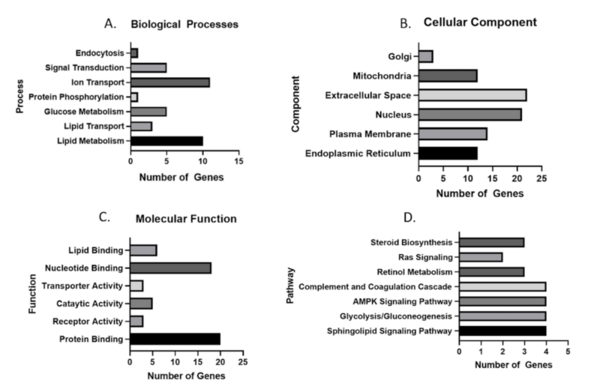
Numerous studies have shown that nonalcoholic fatty liver disease (NAFLD) progression is greatly affected by dysregulation of the hepatic transcriptome. Studies using high throughput technologies such as RNA Sequencing and microarray have identified multiple dysregulated genes in NAFLD. However, these studies utilized vastly different models resulting in few consensus biomarker genes. Thus, in this investigation, we evaluated various datasets to find genes that are similarly expressed across heterogeneous murine models. We hypothesized that there exists a core set of genes dysregulated in nonalcoholic steatohepatitis (NASH), a subtype of NAFLD characterized by steatosis and inflammation. These genes are involved in glucose and lipid metabolism as well as in the inflammatory responses and are the drivers of the observed tissue pathology. To test this hypothesis, we obtained publicly available Gene Expression Omnibus (GEO) datasets from the National Center for Biotechnology Information (NCBI) database and analyzed them to find differentially expressed genes. We also cross-referenced the expression of these genes with published studies for further validation. Our dataset analyses identified 18 genes up-regulated and four down-regulated in at least six of seven datasets. Of these genes, glucokinase (Gck) was up-regulated, and the complement component C8 beta chain (C8b) was down-regulated in every murine dataset analyzed. Both exhibited a similar expression pattern in a human NAFLD dataset. Using approaches such as these, we believe that identifying consistently dysregulated mRNA can lead to the discovery of reliable biomarkers and potentially effective therapeutics in humans despite the heterogeneity of the experimental models used.
This article has been tagged with: