This study explores the link between fingerprints and genetics by analyzing familial fingerprints to show how the fingerprints between family members, and in particular siblings, could be very similar. The hypothesis was that the fingerprints between siblings would be very similar and the dominant fingerprint features within the family would be the same throughout the generations. Fingerprints between the siblings showed a trend of similarity, with only very small differences which makes these fingerprints unique. This work helps to support the link between fingerprints and genetics while providing a modern technological application.
In the United States, there are currently 17.8 million affected by atopic dermatitis (AD), commonly known as eczema. It is characterized by itching and skin inflammation. AD patients are at higher risk for infections, depression, cancer, and suicide. Genetics, environment, and stress are some of the causes of the disease. With the rise of personalized medicine and the acceptance of gene-editing technologies, AD-related variations need to be identified for treatment. Genome-wide association studies (GWAS) have associated the Filaggrin (FLG) gene with AD but have not identified specific problematic single nucleotide polymorphisms (SNPs). This research aimed to refine known SNPs of FLG for gene editing technologies to establish a causal link between specific SNPs and the diseases and to target the polymorphisms. The research utilized R and its Bioconductor packages to refine data from the National Center for Biotechnology Information's (NCBI's) Variation Viewer. The algorithm filtered the dataset by coding regions and conserved domains. The algorithm also removed synonymous variations and treated non-synonymous, frameshift, and nonsense separately. The non-synonymous variations were refined and ordered by the BLOSUM62 substitution matrix. Overall, the analysis removed 96.65% of data, which was redundant or not the focus of the research and ordered the remaining relevant data by impact. The code for the project can also be repurposed as a tool for other diseases. The research can help solve GWAS's imprecise identification challenge. This research is the first step in providing the refined databases required for gene-editing treatment.
Coronary heart disease (CHD) is the leading cause of death in the U.S., responsible for nearly 700,000 deaths in 2021, and is marked by artery clogging that can lead to heart attacks. Traditional prediction methods require expensive clinical tests, but a new study explores using machine learning on demographic, clinical, and behavioral survey data to predict CHD.
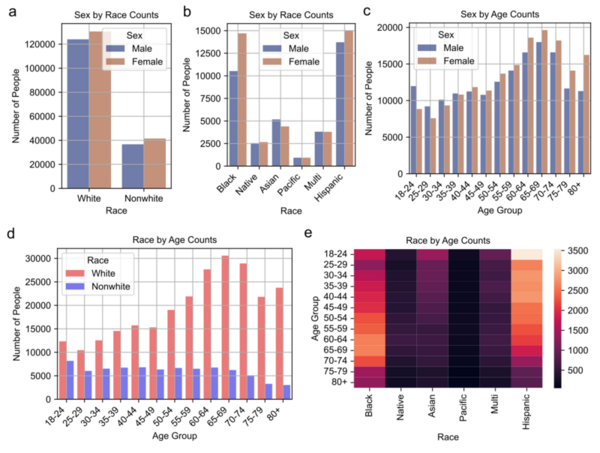
Autism Spectrum Disorder (ASD) and Alzheimer's Disease (AD) are distinct conditions, but research suggests a link, as individuals with ASD are 2.5 times more likely to develop AD. A study employing genome-wide association studies and Mendelian randomization revealed shared genetic factors, particularly in synaptic regulation pathways, that may increase the risk of AD in those with ASD. These findings provide insights into the genetic underpinnings connecting the two disorders.
Although the United States maintains millions of square kilometers of nature reserves to protect the biodiversity of the specimens living there, little is known about how confining these species within designated protected lands influences the genetic variation required for a healthy population. In this study, the authors sequenced genetic barcodes of insects from a recently established nature reserve, the Southwestern Riverside County Multi-Species Reserve (SWRCMSR), and a non-protected area, the Mt. San Jacinto College (MSJC) Menifee campus, to compare the genetic variation between the two populations. Their results demonstrated that the midge fly population from the SWRCMSR had fewer unique DNA barcode sequence changes than the MSJC population, indicating that the comparatively younger nature reserve's population had likely not yet established its own unique genetic drift changes.
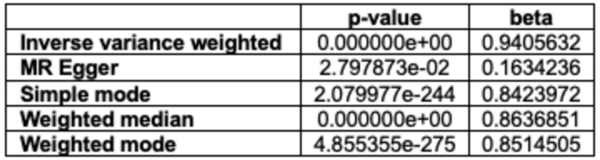
The density of stomata, or stomatal index, in plant leaves is correlated with the plant's rate of photosynthesis, and affected by the plant's climate. In this paper, authors measure the stomatal index of five plant species to derive their rates of photosynthesis. These results could help track changes in plants' photosynthetic rates with changing climate.
Here, seeking to identify a possible explanation for the more frequent diagnosis of autism spectrum disorder (ASD) in males than females, they sought to investigate a potential sex bias in the expression of ASD-associated genes. Based on their analysis, they identified 17 ASD-associated candidate genes that showed stronger collective sex-dependent expression.
Here the authors sought to understand the effects of different variables that may be tied to pollution and climate change on genetic variation of Pacific white-sided dolphins, a species that is currently threatened by water pollution. Based on environmental data collected alongside a genetic distance matrix, they found that ocean currents had the most significant impact on the genetic diversity of Pacific white-sided dolphins along the Japanese coast.
In this study, the authors developed a model named DNA Sequence Embedding Network (DNA-SEnet) to classify DNA-asthma associations using their genomic patterns.
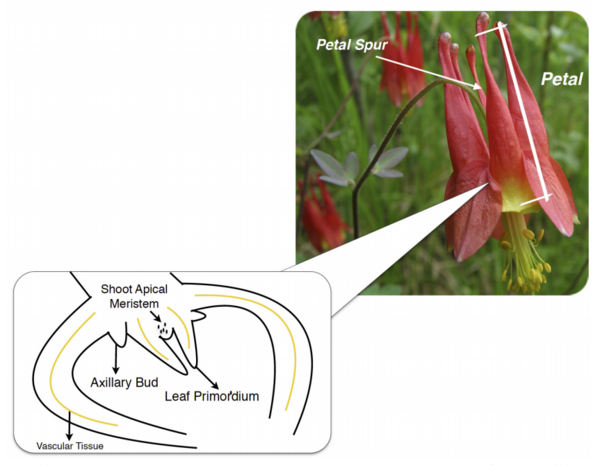
Plants, and all other multi-cellular organisms, develop through the coordinated action of many sets of genes. The authors here investigate the genes, in a class named KNOX, potentially responsible for organizing a certain part of Aquilegia (columbine) flowers called petal spurs. Through the technique Reverse Transcription-Polymerase Chain Reaction (RT-PCR), they find that certain KNOX genes are expressed non-uniformly in petal spurs, suggesting that they may be involved, perhaps in a cell-specific manner. This research will help guide future efforts toward understanding how many beautiful flowers develop their unique shapes.