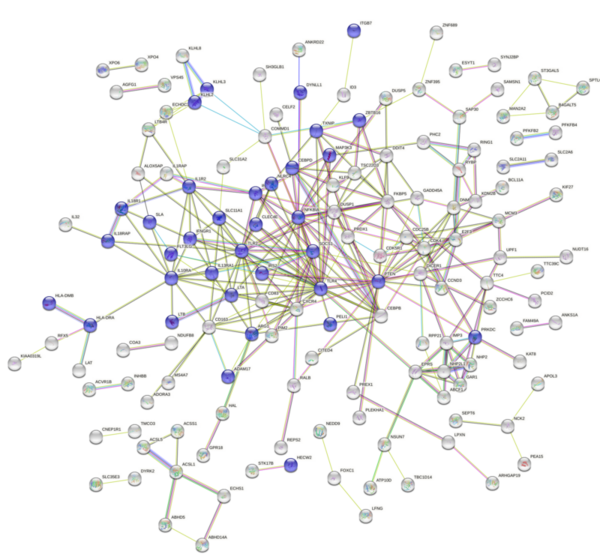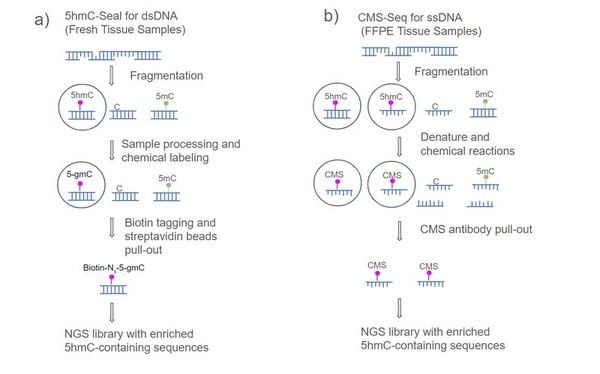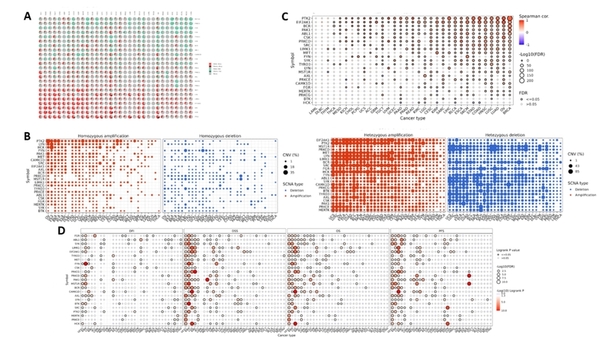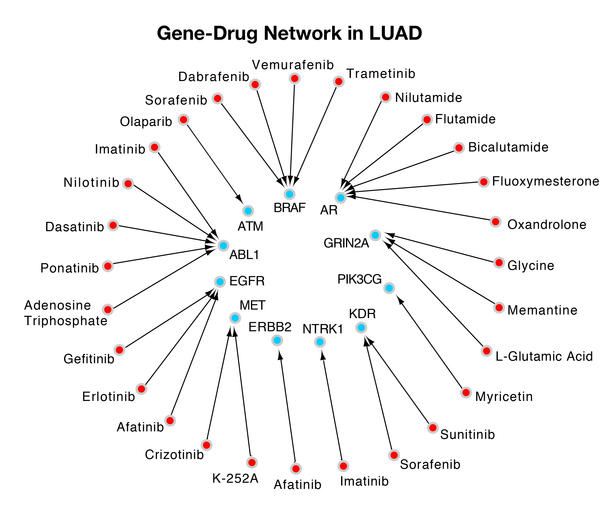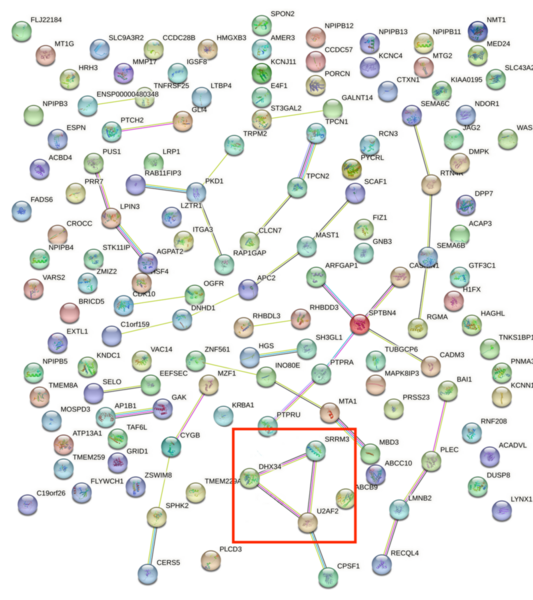
Childhood abuse has severe and lasting effects throughout an individual's life, and may even have long-term biological effects on individuals who suffer it. To learn more about the effects of abuse in childhood, Li and Yearwood analyze gene expression data to look for genes differentially expressed genes in individuals with a history of childhood abuse.
Read More...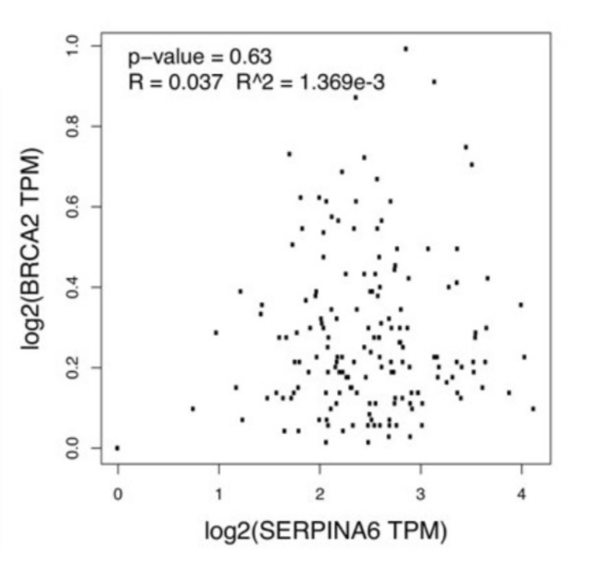
%20(1).png)