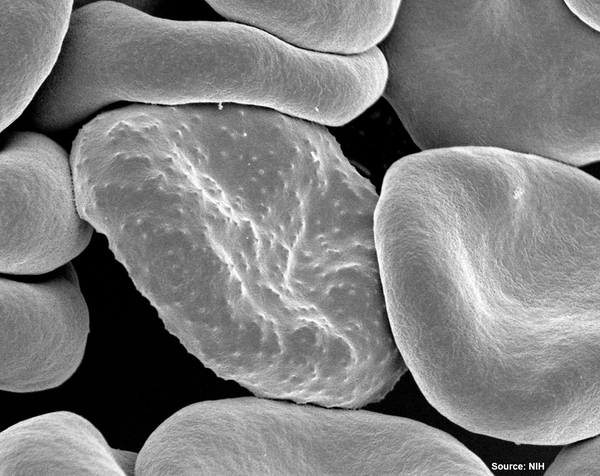
The diagnosis of malaria remains one of the major hurdles to eradicating the disease, especially among poorer populations. Here, the authors use machine learning to improve the accuracy of deep learning algorithms that automate the diagnosis of malaria using images of blood smears from patients, which could make diagnosis easier and faster for many.
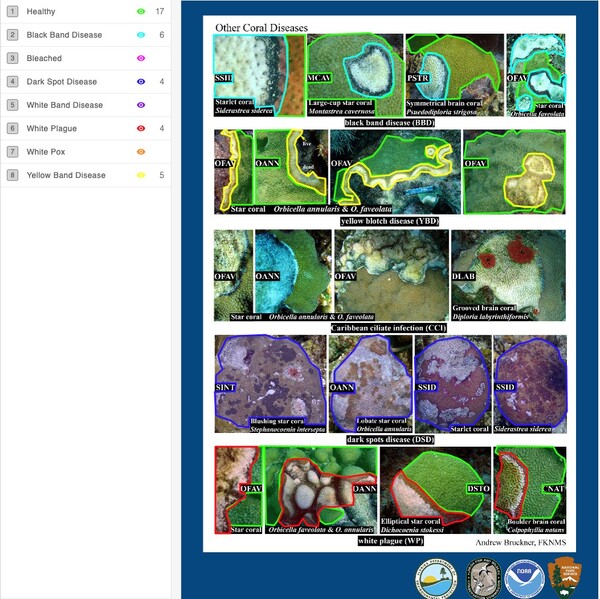
Triggered largely by the warming and pollution of oceans, corals are experiencing bleaching and a variety of diseases caused by the spread of bacteria, fungi, and viruses. Identification of bleached/diseased corals enables implementation of measures to halt or retard disease. Benthic cover analysis, a standard metric used in large databases to assess live coral cover, as a standalone measure of reef health is insufficient for identification of coral bleaching/disease. Proposed herein is a solution that couples machine learning with crowd-sourced data – images from government archives, citizen science projects, and personal images collected by tourists – to build a model capable of identifying healthy, bleached, and/or diseased coral.
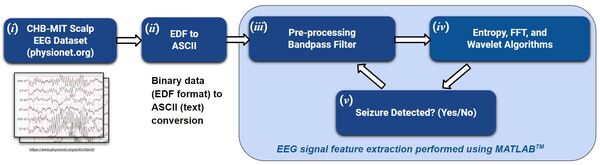
Each year, over 100,000 patients die from Sudden Unexpected Death in Epilepsy (SUDEP). A reliable seizure warning system can help patients stay safe. This work presents a comprehensive, comparative analysis of three different signal processing algorithms for automated seizure/ictal detection. The experimental results show that the proposed methods can be effective for accurate automated seizure detection and monitoring in clinical care.
Although the United States maintains millions of square kilometers of nature reserves to protect the biodiversity of the specimens living there, little is known about how confining these species within designated protected lands influences the genetic variation required for a healthy population. In this study, the authors sequenced genetic barcodes of insects from a recently established nature reserve, the Southwestern Riverside County Multi-Species Reserve (SWRCMSR), and a non-protected area, the Mt. San Jacinto College (MSJC) Menifee campus, to compare the genetic variation between the two populations. Their results demonstrated that the midge fly population from the SWRCMSR had fewer unique DNA barcode sequence changes than the MSJC population, indicating that the comparatively younger nature reserve's population had likely not yet established its own unique genetic drift changes.
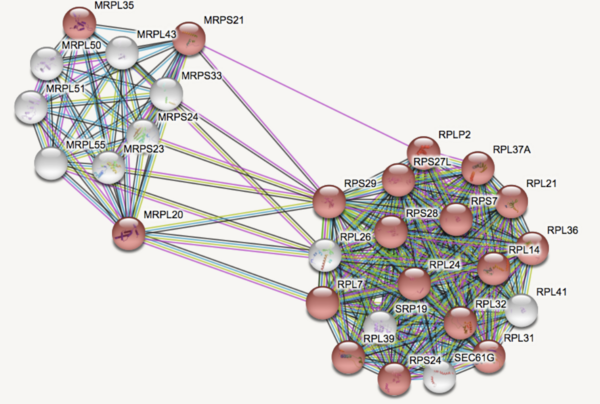
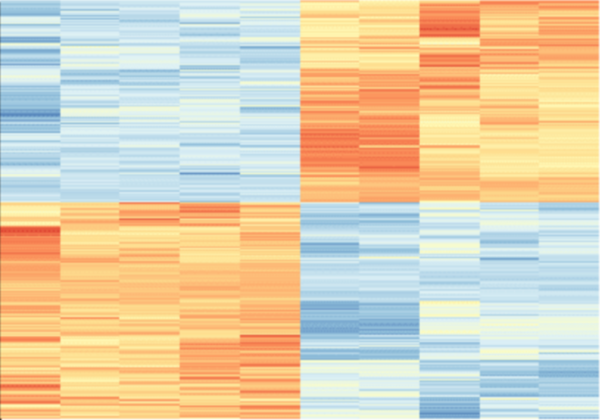
Major Depressive Disorder (MDD), and Post-Traumatic Stress Disorder (PTSD) are two of the fastest growing comorbid diseases in the world. Using publicly available datasets from the National Institute for Biotechnology Information (NCBI), Ravi and Lee conducted a differential gene expression analysis using 184 blood samples from either control individuals or individuals with comorbid MDD and PTSD. As a result, the authors identified 253 highly differentially-expressed genes, with enrichment for proteins in the gene ontology group 'Ribosomal Pathway'. These genes may be used as blood-based biomarkers for susceptibility to MDD or PTSD, and to tailor treatments within a personalized medicine regime.
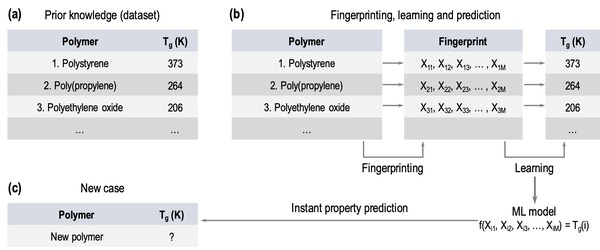
In this study, the authors test whether providing a larger dataset of glass transition temperatures (Tg) to train the machine-learning platform Polymer Genome would improve its accuracy. Polymer Genome is a machine learning based data-driven informatics platform for polymer property prediction and Tg is one property needed to design new polymers in silico. They found that training the model with their larger, curated dataset improved the algorithm's Tg, providing valuable improvements to this useful platform.
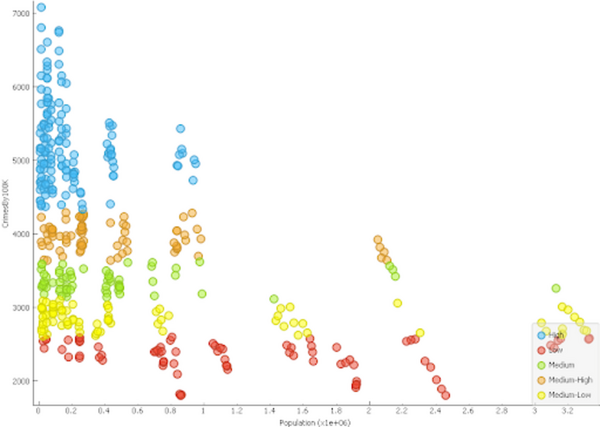
The authors look at using publicly available data and machine learning to see if they can develop a criminal activity index for counties within the state of California.
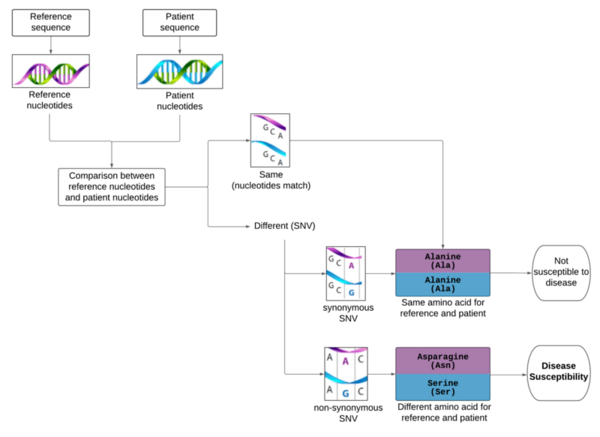
In the United States, there are currently 17.8 million affected by atopic dermatitis (AD), commonly known as eczema. It is characterized by itching and skin inflammation. AD patients are at higher risk for infections, depression, cancer, and suicide. Genetics, environment, and stress are some of the causes of the disease. With the rise of personalized medicine and the acceptance of gene-editing technologies, AD-related variations need to be identified for treatment. Genome-wide association studies (GWAS) have associated the Filaggrin (FLG) gene with AD but have not identified specific problematic single nucleotide polymorphisms (SNPs). This research aimed to refine known SNPs of FLG for gene editing technologies to establish a causal link between specific SNPs and the diseases and to target the polymorphisms. The research utilized R and its Bioconductor packages to refine data from the National Center for Biotechnology Information's (NCBI's) Variation Viewer. The algorithm filtered the dataset by coding regions and conserved domains. The algorithm also removed synonymous variations and treated non-synonymous, frameshift, and nonsense separately. The non-synonymous variations were refined and ordered by the BLOSUM62 substitution matrix. Overall, the analysis removed 96.65% of data, which was redundant or not the focus of the research and ordered the remaining relevant data by impact. The code for the project can also be repurposed as a tool for other diseases. The research can help solve GWAS's imprecise identification challenge. This research is the first step in providing the refined databases required for gene-editing treatment.
Although the 5-year survival rate for colorectal cancer is below 10%, it increases to greater than 90% if it is diagnosed early. We hypothesized from our research that analyzing non-synonymous single nucleotide variants (SNVs) in a patient's exome sequence would be an indicator for high genetic risk of developing colorectal cancer.