Analysis of the lung microbiome in cystic fibrosis patients using 16S sequencing
(1) Basis Independent Silicon Valley, San Jose, California, (2) Department of Bioengineering, Stanford University, Palo Alto, California
https://doi.org/10.59720/22-234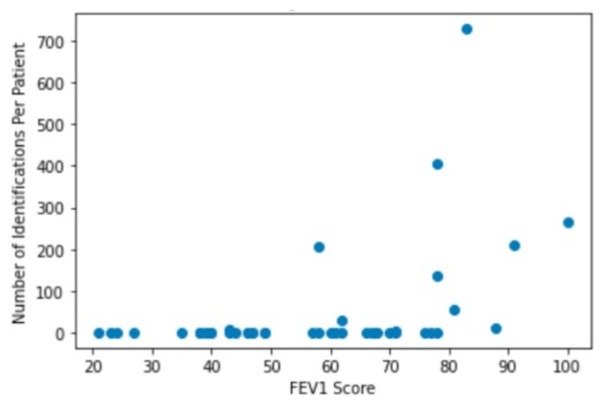
Cystic fibrosis (CF) patients often develop lung infections that range anywhere in severity from mild to life-threatening due to the presence of thick, sticky mucus that fills their airways. Since many of these infections are chronic, they not only affect a patient’s ability to breathe but also increase chances of mortality by respiratory failure. Examining the lung microbiology of CF patients can help with the prediction of the current condition of lung function, with the potential to guide doctors when designing personalized and thereby more effective treatment plans for patients. With a publicly available dataset of DNA sequences from bacterial species in the lung microbiome of CF patients, we investigated the correlations between different microbial species in the lung and the extent of deterioration of lung function. 16S sequencing technologies allowed us to determine the microbiome composition of the samples in the dataset. We found that the Fusobacterium, Actinomyces, and Leptotrichia microbial types all had positive correlation with forced expiratory volume in 1 second (FEV1) score, indicating potential displacement of these species by pathogens as the disease progresses. However, the dominant pathogens themselves, including Pseudomonas aeruginosa and Staphylococcus aureus, did not have statistically significant negative correlations with FEV1 score as described in past literature.
This article has been tagged with: